
How does arthritis actually work?
The immunology of rheumatoid arthritis. Tracing an experimental campaign, looking at current treatments and exploring mechanisms for new drugs.

Arthritis is a common and painful disease, but we still don’t know exactly how it starts or progresses with molecular precision. Somewhat simple symptoms emerge from an intricate web of cells and molecules with strong interactions with the immune system. It is a fascinating case study in complex disease.
In this essay, we will dive into a series of immunology experiments that show a rare population of T cells get stuck in flaring joints and recruit circulating immune cells from across the body to cause the characteristic pain, stiffness and swelling. We then look at approved treatments for the disease, why they might not work that well, and explore alternative routes for new drugs. Along the way, we explore clever methods in immunology that allow us to engineer and introspect the immune systems of organisms.
Arthritis is not one disease
Its a broad group of different diseases that all cause inflammation and damage in the joints. While one in four Americans have arthritis, the vast majority (80%) have a type known as osteoarthritis, caused by the mechanical “wear and tear” of joints primarily from overuse in sports or obesity.
A smaller number, but still a very large group (1% of Americans), have rheumatoid arthritis (RA), an autoimmune flavor that’s more unpredictable and dispersed in joints across the body. RA will be the focus of this essay. In contrast to osteoarthritis, the joint pain is generally independent from activity and manifests in random, periodic flares of pain with long periods of remission. A lot of people have RA. It's likely you know several or have it yourself.
A rare population of T cells might cause arthritis
In 2021, a group of researchers out of Boston Children’s Hospital published a series of experiments suggesting a rare population of T cells lodge themselves in joint tissue in RA patients. Their data shows the specific joints where the arthritic flares show up are the same joints where these cells are lodged. They also showed these cells do not directly cause the flares. Rather, immune cells from throughout the body are recruited to these joints by the lodged T cells, where they are coaxed into various inflammatory states leading to the characteristic pain, stiffness and swelling.
While not conclusive, this paper suggests this T cell population plays a central, perhaps causal, role in the manifestation of this disease. This paves the way for better precision therapies and maybe durable cures (eg. by somehow get rid of the lodged cells). This is exciting. There is no cure for RA and most patients take multiple medicines for the rest of their life to curb symptoms.
This paper is also a great case study in experimental design and understanding how broad scientific questions are broken down into campaigns of smaller experiments. Using a range of different molecular assays and disease models, the researchers systematically tossed out plausible hypotheses and whittled away at progressively deeper drivers of disease. We’ll dive into this chain of experiments and learn about interesting techniques in immunology along the way.
Tracing an end-to-end immunology campaign
We begin this essay by following the in-vivo work from Chang et al. in some detail. The high level steps are as follows:
Creating models of RA in mice
Using adoptive transfer to determine the unique role of CD4 and CD8 T cells in RA
Identifying a rare T cell subtype with flow cytometry
Further characterizing this rare cell population by their actual movement in tissue
Showing this population itself does not cause flares but recruits other immune cells
Validating what we can with human data
This is an immunology study, so of course we begin our journey in mice. We’ll approach our mouse data with latent skepticism, as we should with all translational research. We will later validate some of the findings in people, but first we apply a host of interesting engineering techniques to create, control and pick apart the molecular drivers of arthritis in these organisms.
How to model arthritis in a mouse
Constructing a mouse model of RA is surprisingly straightforward. We inject an antigen into the wrist, knee and ankle on a single side of its body. The opposing joints are injected with a salt buffer as a control.
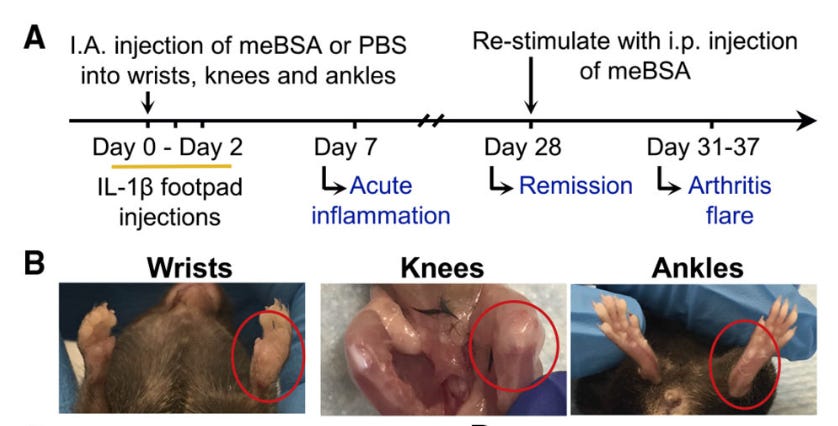
The antigen injected joints quickly swell up, but after a month they return to a “normal” state. Consistent with the periodic arthritic flares in RA patients, the swelling can be induced by systemic, not joint localized, injection of more of the same antigen into the chest of the mouse. This is actually somewhat surprising. It is not obvious that injecting the antigen somewhere else will cause the “primed” joints to flare up in this way.
It is important to pause and consider why this is a sufficient model of RA:
The mice have joint specific memory. The arthritic flares reoccur in the same joints that were initially challenged. They don’t occur elsewhere. Simple, but consistent with the disease in people.
The flares lie dormant for long periods of time before sparking up again. Another key property of RA.
The injected joint tissues have lots of immune cells in them. If we sacrifice these diseased mice and break apart their joints, we can identify familiar immune cells using flow cytometry - B cells, macrophages, neutrophils and, importantly, T cells.
Animal models are imperfect. Mice literally have different immune systems than people, with analogous cell types that display different cell markers and release distinct sets of chemokines/cytokines. Yet, these distinct properties give us some confidence in disease recapitulation and suggest there is more we can learn.
Why even look at T cells in arthritis?
Around the time this study was published, there was a good deal of genetic evidence suggesting CD4 (“helper”) T cells were involved in the disease. The conclusion articulates this:
“T cell studies in RA have focused primarily on CD4+ T cells because … RA is strongly associated with the major histocompatibility complex class II (MHC class II) HLA-DRB1 alleles”
Which means that RA patients are more likely than the average person to display specific variants of MHC II, the surface proteins central to the activation of CD4 T cells (link). Indeed, at this time there was some work showing CD4 T cells might encourage B cells to produce autoantibodies that cause the flares.
However, this same section also sheds some light on some seemingly less understood genetic associations with the MHC I, or CD8 (“killer”) T cell associated receptors, that might have sparked this entire study:
"... however RA also exhibits genetic associations with alleles in the MHC class I HLA-B locus, highlighting the likely importance of CD8+ T cells.”
While CD8 T cells were described as “being around” (actually way back in 2002), their precise role had yet to be explored.
Using adoptive transfer to zoom in on killer T cells
Adoptive transfer is a technique in immunology where well understood, often engineered, cells are transferred from one organism to another. The transferred cells have distinct surface markers (in this case transgenic CD45.1) so they can be filtered out and characterized in the resulting immune response.

The transferred T cells are clonally restricted; they express only one type of TCR (healthy mice have ~10 million distinct T cell clones). This means they should all respond precisely to a single antigen that we can deliberately introduce and shouldn’t otherwise be there. While the adaptive immune system of the mouse can kick in and build new T cells that are also specific to this antigen, the transferred cells should be present in much greater numbers, making them easy to study.
For our purpose, we can take CD8 and CD4 T cells restricted to recognize only ovalbumin from an engineered animal and transfer them into the mouse model of RA we described above. After giving the new mouse ovalbumin, we can break apart the flaring joints and see which of the two T cell types are still there.
Here we find our first really interesting result: both T cell types, CD4 and CD8, are present during the flares.

Yet only the CD8 T cells stick around during the period between flares. Why?
A special type of T cell lodge themselves in arthritic joints
An interlude on cell types
The immunology literature classifies the “types” (and subtypes and sub-subtypes) of various immune cells with an obsession and attention to detail rivaled only by Victorian-era taxonomists and modern botanists. While this jargon can be overwhelming to the unindoctrinated, these terms are very functional. Trained immunologists use this shorthand to quickly allude to an iceberg of literature and data beneath each word to avoid repetition.
The CD4/CD8 T cell groups merely scratch the surface. Each helper T cell can be organized by one of three major immune modules it participates in: Th1 (pronounced “T helper 1”), Th2, and Th17. These can be further binned by a naive, activated, memory or exhausted state, among others, which themselves are also broad descriptions of behavior. A “gastroepithelial troping, activated, but slightly exhausted, T helper 17 cell” is something you might read about in an immunology paper (and in fact might be involved in celiac disease).

The focus of the remainder of these experiments, and the putative driver of RA in these models, is a small population of CD8 T cells called “tissue resident memory CD8 T cells”. Both the “tissue resident” and “memory” states can exist independently and are important to understand.
Unlike most T cells, which circulate freely throughout the body, tissue resident T cells lodge themselves in a single place and stay put. In healthy people, they serve as a local surveillance mechanism that rapidly clears repeat pathogens, especially those with preference for certain tissues (Salmonella in the gut, tuberculosis in the lungs, hepatitis in the liver).
In contrast, memory T cells describe a state in the developmental cycle of these immune cells. After developing in the bone marrow, early “naive” T cells are born in the thymus before circulating around the body looking for invaders. If they encounter a suitable pathogen, they might become “active”, but many of them quickly die off soon after the pathogen is cleared. A small fraction of these T cells become “memory” cells and persist in a dormant state awaiting more pathogens.

So our “tissue resident memory CD8 T cells” (hereafter referred to as T_RM) are cytotoxic T cells that lodge themselves in the joints and have memory for past antigen.
How do we find these T_RM cells?
We previously showed that CD8 T cells stick around in between flares. Now we must show the T_RM subset does the same. We do the same thing as before - inject the mouse in its joints, wait a bit and inject it again in its chest - but now we break apart the joints and use flow cytometry to look at various T cell marker genes.

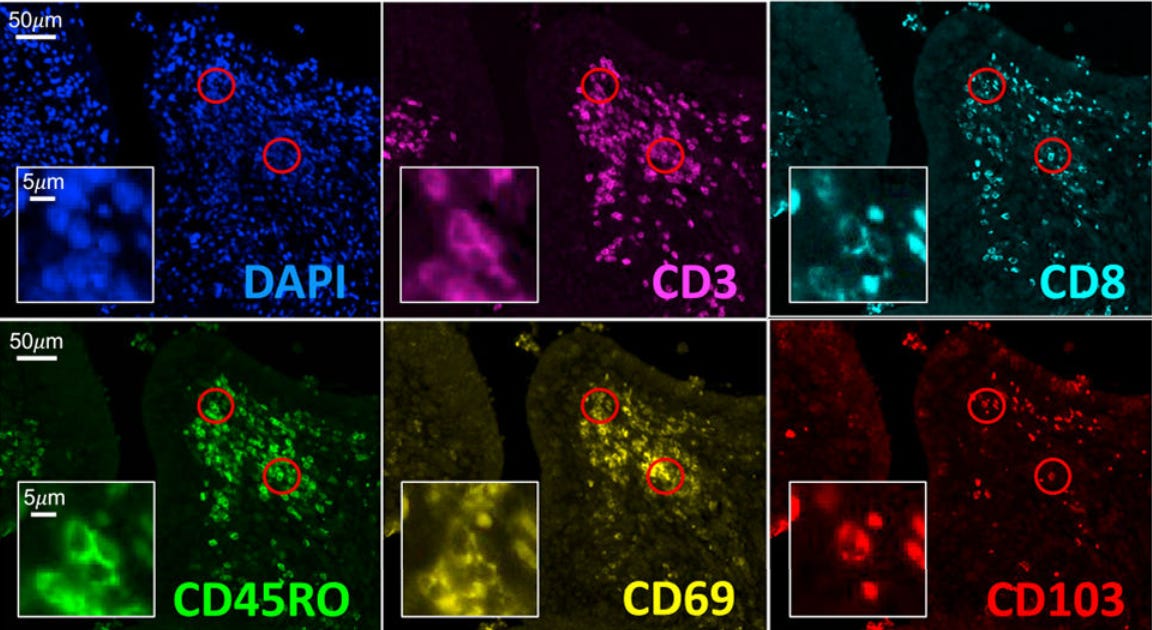
When we do this, we find a unique population of cells, present only in the treated joints (not the controls), that display the “established” markers of the T_RM type - CD8, CD44, CD69, CD103, among others. CD44 is an “experience marker” that tells us the cell has previous exposure to antigen. CD103 is another important one: a sticky integrin that binds to epithelial tissue, locking its T cell in place.
We can do better than look at just the surface proteins and actually track the position of these cells over time to confirm they stay put. By injecting an AAV loaded with a recombinase payload into the flaring joints, we can label the T cells with a fluorescent marker and check back after a month to see if the labeled cells are still there. Indeed they are.

But we can do more. True T_RMs are oblivious to chemical signals that pull normal T cells out of the tissue and back into circulation. CCL21 is a major one that normally rallies them back to the lymph nodes. If we pull our T cells out of the joints, put them on one side of a chamber, and put CCL21 on the opposite side of this same chamber, we might expect them to migrate to the CCL21. However, in practice, the same small population expressing CD8, CD44, CD69 and CD103 stay put in the chamber after a few hours, showing their sensitivity to CCL21 in true tissue resident fashion.

Layering these different experiments builds a holistic picture of the cells and increases our confidence they really are T_RMs. Remember, the laundry list of “markers” are a quick and dirty way to identify *behavior*, but the behavior itself is what is truly important. Supplementing the biochemical signatures with how they actually move is a cool way to verify this.
But T_RMs don’t actually cause the flares?
Now we arrive at our second very interesting piece of information. Despite T_RMs playing a large role in RA flares, it’s likely they don’t cause the pain and swelling directly, but rather recruit a host of other immune cells from across the body that cause the joint inflammation.
As we briefly saw with CCL21, lymphocytes follow trails of chemical gradients around the body. Their “movement” is really more of a random walk: a series of random molecular sampling events than any real mechanical motion. We can shut off this random walk by getting rid of the chemicals composing these gradients (SIP and CXCL5) and preventing the flood of lymphocytes to the joints.

When we do this, the flares stop, even though the T_RMs are still there and even expanding in number.
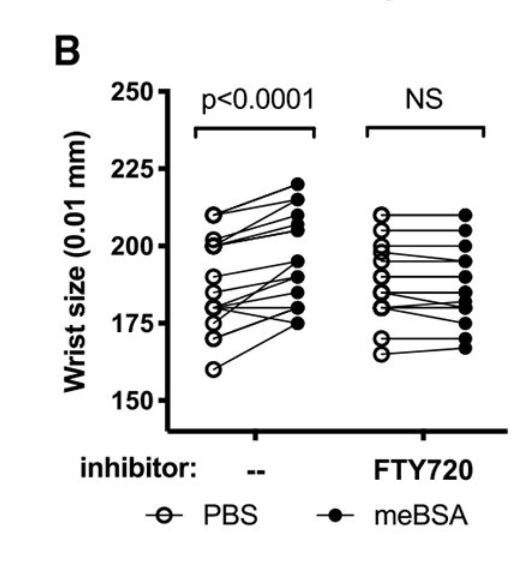
We can also test the opposite. We remove the T_RMs lodged in the joint and allow the circulating lymphocytes to flow around the body freely. After using another recombinase to selectively kill cells in the joints during remission (there are different cool systems to do this), and allowing the peripheral T cells to flow freely, we inject antigen into the chest of the mice.

Once again, the flares are gone. Immune cells, in the absence of T_RMs in the joints, don’t cause arthritic flares. At least in mice.
How much of this translates to people?
The authors from this paper collected a bunch of human RA joint samples and looked at some single cell data + flow data. They found similar protein and transcriptional profiles characterizing T_RM cells.
They explored orthogonal public datasets and found similar T_RM gene signatures.
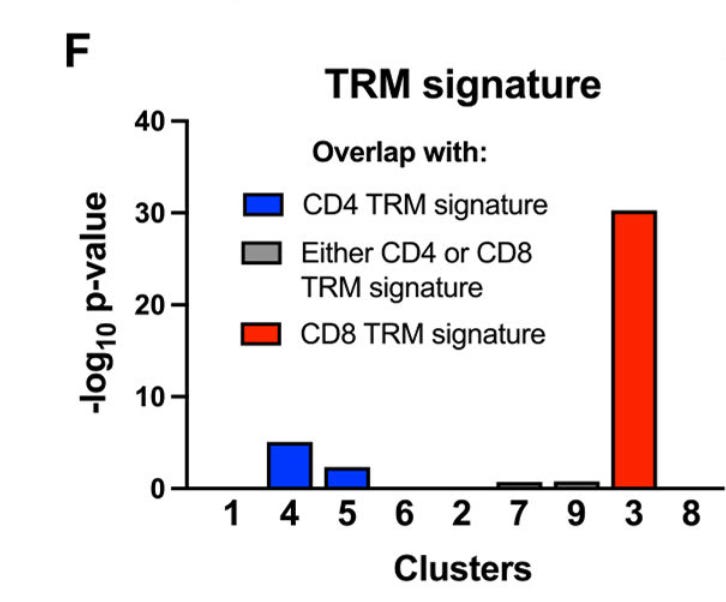
A curious piece of new information was a restricted TCR repertoire in this human transcriptional data, perhaps indicating the memory of antigen challenge in T_RM.
However, holistic translational work was lacking. No experiments exploring the specific behavior of the lodged cells - their role in recruiting other immune cells or their response to chemokines - were performed. This makes sense for the scope of the paper but should temper truly durable conclusions about the role of this new information in driving human RA.
Furthermore, a cursory glance over the RA literature reveals other drivers at play: synovial macrophages, aggressive fibroblasts, and autoantibodies. While the immune recruitment could explain each of these, it also might not. The only way to know for sure is to test it.
Turning this information into medicines
Diseases are great ways to learn about new biology, particularly because you really begin to understand the utility and intricate engineering of the molecular components that break. However, the truly exciting promise of basic research lies in the generation of new knowledge that can be used to make drugs that save or change lives.
In this second section, we first look at how RA is treated and why each group of drugs kind of works but not entirely. We then use information from the study to come up with plausible new drugs.
How current drugs work
There are several classes of drugs approved to treat RA, ranging from non-specific inflammation modulators (ibuprofen) to very precise engineered antibodies (the lauded HUMIRA). We’ll speedrun through some of these therapeutic classes, look at their mechanisms and use our new information to think about why they might work or not work for RA. Fair disclaimer: this is all pedagogical speculation.
Nonsteroidal Anti-Inflammatories, Steroids + Conventional Small Molecules
Nonsteroidals (eg. Ibuprofen), glucocorticoids (eg. Prednisone) and “conventional” small molecule treatments (eg. Hydroxychloroquine) generally all reduce inflammation by targeting common pathways most cells in your body share.
 Inhibits the Antithrombotic Activity of Aspirin")
They are not very precise. It is as easy to see why they help curb the symptoms of RA - the swelling, stiffness and pain - and just as easy to see why they leave room for improvement.
Targeted Small Molecules (JAK Inhibitors)
Many behaviors of the T cell (and most immune cells) rely on chemokines and cytokines that signal through a group of intracellular chemical pathways sharing JAK/STAT proteins.

We explored some of these JAK/STAT mediated functions: the initial “stickiness” to the joint tissue by T_RMs, the recruitment of additional immune cells, the inflammation from the recruited cells. Each of which could be switched off if the correct JAK/STAT complex is knocked out. JAK inhibitor drugs (eg. Tofacitinib, Baricitinib, Upadacitinib, Filgotinib) do exactly this.
There are a few reasons why these inhibitors, commonly used today to treat RA, “kind of work”:
T_RMs recruit circulating immune cells using chemokines, like CCL5. We even walked through an experiment showing that blocking CCL5 prevented T cell recruitment and dropped flares.
The inflammation responsible for “flares” almost certainly comes from effects of cytokines on the recruited immune cells, not T_RMs. There are many different inflammatory programs involved, each regulated by different JAK/STAT pathways.
Despite being systemic treatments, not specific to joints, data shows JAK inhibitors are effective at reaching tissue resident T cells (at least in other tissue contexts).
But they also don’t “work all the way”:
JAK inhibition will not dislodge the T_RM from the tissue it inhabits, so it continues to signal for fresh and varied immune cells, over and over again, perhaps why lifetime treatment is necessary.
There are also many variants of JAK, each associated with distinct JAK/STAT pathways. These variants are involved in different chemokine axes, each distinct in structure but similar in function to CCL5, that can recruit other immune cells.
There are different variants in the three broad classes of inflammatory immune modules, of which some subset is recruited by T_RM.
So the theme here is JAK inhibitors likely work by indirectly and partially removing inflammatory programs in a subset of recruited immune cells.
Targeted Biologics
Monoclonal antibodies, engineered to bind to a range of immune targets with great precision, are also common treatments for RA. There are an incredible number of approved antibody therapies and we can group them by what they bind to.
TNF-α binders, like Infliximab, Adalimumab, Etanercept, Golimumab, target broad programs of inflammatory behavior, certainly shared with other cells and healthy immune function.

IL-6 is necessary to differentiate CD4 T cells into the Th17 (from the so-called “type 3 immune program” we briefly discussed). It’s actually well understood that Th17 cells play a large role in RA progression. IL-6 antibodies, like Tocilizumab, Sarilumab, likely deplete these cells.
IL-1 targeting antibodies prevent breakdown of bone, cartilage and rampant fibroblast growth (among many other general immune things). Anakinra is one such binder.
There is a clear theme here. Each successful (or unsuccessful) monoclonal precisely targets an imprecise component of the disease. It is likely none of these inflammatory pathways, immune cell fate commitment programs or other immunology “things” are the cause of RA. Rather, as in the JAK inhibitor case, they are part of the problematic soup of immune cells that are drawn into the joints by our T_RM population. Because none of these drugs dislodge the T_RMs, they also don’t cure the flares.
This also helps explain the variability between patient populations in drug response. Even if the T_RM cells are shared amongst all RA patients, the recruited immune population will certainly be different, due to natural immunological variation in people. Some might have a lot of Th17 cells and respond well to IL-6 blockers, while others might suffer from invading neutrophils and respond to HUMIRA (adalimumab).
Can we repurpose existing drugs using this information?
I was unable to find existing programs that focus on T_RM, either through a direct claim or unambiguous molecular target. That certainly doesn’t mean such development efforts do not exist. What I was able to do was come up with some reasonable mechanisms for new drugs that integrate specific new information from this paper. Some of these mechanisms might even be able repurpose existing drugs.
Blocking CCL5 signaling
Recall CCL5 is one of the important chemical trails leading circulating immune cells into the joints. We just showed that if we disable it, the flares go away (in mice). There are actually several drugs that block the interaction of CCL5 with its receptor CCR5.
Leronlimab is a CCR5 blocking monoclonal currently in development for HIV, with recent moves to also try the drug in chronic inflammation.
Cenicriviroc is a CCR5 blocking small molecule, also in development in HIV and recently failed to treat NASH.

Interestingly, Maraviroc, a small molecule CCR5 antagonist approved for HIV failed to show efficacy for RA in 2012. This need not diminish confidence in this mechanism as eg. issues with binding kinetics in new cell type populations, lack of synovial tissue penetration or other factors could be at play. But the lymphocyte trafficking could also occur along different chemokine axes. The CCL5 blocking data was generated in a mouse after all.
Preventing the T_RMs from “sticking”
The sticky integrin α_Eβ_7 (CD103) is likely a core structural component to tissue residency. We also saw that the synovial T_RM identified in the paper definitely expresses CD103 (in mice and people). There are no FDA approved drugs that target this variant of integrin, there are clinical successes for other variants.

An anti-α_Eβ_7 (CD103) approach in RA either remains preclinical in stealth, or doesn’t exist altogether, but might make a lot of sense given the precision with which it would target T_RM cells.
Spot removal of the T-cells in the joints
Depletion of T cells with local administration of broad acting drugs, like anti-CD3 antibodies (Muromonab-CD3) used with transplant patients, might be the most convincing path forward. Site specific depletion is known to cause systemic immunosuppression or cytokine release syndrome, however, the joint is a pretty small chunk of tissue and conducive to spot injection with small amounts of drug. Excited to see activity here over the next few years if this makes sense.
We will cure RA with human ingenuity
One goal of this exercise is to expose scientists and engineers to new immunology techniques and biology. However, another goal is to highlight how tractable the path forward to curing complex disease like RA is.
The problem is soluble with enough clever human minds designing experiments, capital to run trials and time to see the effects of new drugs in people. This is not to diminish the complexity or difficulty of drug development, but to show that these diseases are not magic black boxes impenetrable by the scientific method. With the exponential improvement (in cost + throughput) of molecular assays, better translational models and exciting new classes of therapies, understanding and curing RA will likely happen in our lifetime. This will only happen faster with more smart people working on the problem.
LatchBio is a team of engineers building data infrastructure to accelerate bioengineering. We are thinking deeply about the software needed to understand experimental campaigns like the one described, both to coordinate diverse scientific teams and synthesize information across experiment types.
My partner and I literally asked this question yesterday and were hoping to understand the mechanisms. Great piece!