
Modern Infrastructure for Bulk RNA-seq
structured metadata capture, hosted nf-core/rnaseq, programmable dashboards to discover genes + function, collaborative Jupyter / RStudio environments, Illumina + AWS + GCP integrations

Despite the rise of single cell and spatial transcriptomics, bulk RNA-seq continues to be the workhorse assay for most of the biotech industry. It is cheap, easy to run in the lab and well understood.
However, the data infrastructure for the storage, analysis and reporting of results is often lacking for this experiment. Disproportionate attention is placed on the bioinformatics workflows that process sequencing reads into gene counts, at the expense of the upstream and downstream infrastructure necessary for collaboration, compliance and future analysis.
This includes:
Central and structured capture of sequencing data and experimental metadata
Visualizations and reports that are accessible and usable by scientists
Links between raw experimental data, bioinformatics workflows, processed counts and final reports
Bulk RNA-seq analysis is as much a human engineering project as a software problem and demands infrastructure where each component is centralized and accessible to different wet and dry lab teams. Here we present the full analysis lifecycle of this experiment on LatchBio to answer the following biological questions:
What are the gene counts in my sample?
What are the differentially expressed genes between conditions?
What are the key functions, pathways or ontologies associated with key genes or gene sets?
Integrate upstream data sources
The analysis life cycle begins with a central storage location with direct links to sources of sequencing data. Latch Data streams sequencing files from Illumina machines, with automated pulls from BaseSpace, and supports AWS / GCP bucket mounts.
When FastQ files are uploaded, FastQC is automatically run, where a double-click will reveal a quality control report.
The long-term data storage system also provides provenance, providing a clear ledger of upload and download events for any file object, linking the user, bioinformatics workflow or data source when appropriate.
Capture experimental metadata
Structuring raw sequencing outputs with necessary metadata from the wet lab is necessary for flexible downstream analysis and data re-use long into the future. Latch Registry allows platform teams to create typed and error-validated schemas to model their bulk RNA-seq experiments.
This allows bench scientists to provide information about their experimental design, such as cell line, drug dose and growing conditions, in a graphical interface. The schemas link this metadata directly to both raw sequencing files and processed results, like gene counts and downstream reports.
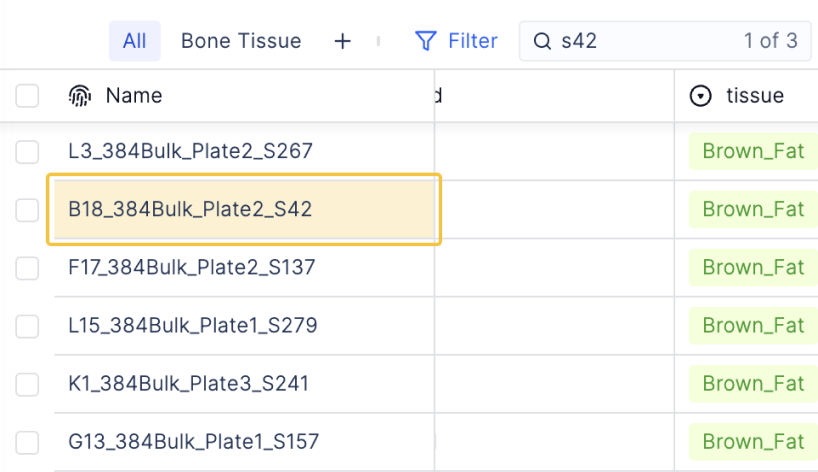
Typed column values, and graphical error validation, preserve a consistent structure for future exploratory analysis.
Generate counts from FastQs with nf-core/rnaseq
nf-core/rnaseq is an open-source bioinformatics workflow that processes raw sequencing reads, aligns them to genes and performs quality control checks. The pipeline uses gold-standard tools and is maintained by the growing nf-core community.

This workflow is hosted on Latch Workflows, using a native Nextflow integration, with a graphical interface for accessible analysis by scientists. It can be modified or extended as needed with minimal modifications to existing Nextflow code.
There is also an integration with Latch Registry so that batched workflows can be launched from “graphical sample sheets” or tables associating raw sequencing files with metadata.
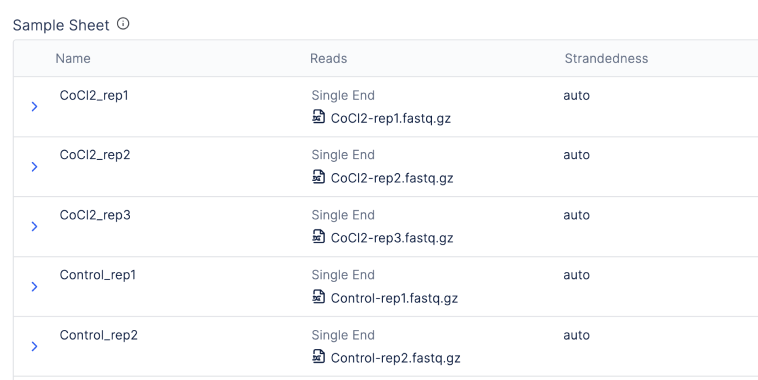
Type-safe and error validated values from scientists are matched with workflow parameters.

The managed computing infrastructure scales to hundreds of samples, with clear logging and error-reporting. Data provenance links versioned and containerized workflow code to input and output files.
Identify differentially expressed genes with DESeq2
Discovering genes that change meaningfully between conditions is often the main goal of bulk RNA-seq analysis.
DESeq2 is a popular tool that accomplishes this by testing statistical differences between gene sets. It is accessible to scientists on Latch Workflows, downstream of nf-core/rnaseq, where they can construct condition groups in a graphical interface and launch their own analysis. The underlying code is accessible and can be downloaded and modified.
Genes with significant changes between conditions are then visualized in downstream dashboards with volcano and MA plots. The underlying Python code that produced these plots can also be accessed and modified.
Discover gene sets, functional annotations, mechanisms with GSEA, KEGG
Identifying pathways, functional descriptions or gene sets associated with significant genes allows scientists to form biological hypotheses around the effect of conditions.
A dashboard on Latch Plots consolidates many of these public databases behind an interface and allows scientists to self-serve the results.
Exploratory analysis in Jupyter / RStudio
Biology is complex and it would be naive to assume that turn-key workflows and pre-built dashboards are sufficient for many projects. In addition to providing access to the underlying code for workflows and plots mentioned, Latch Pods allows computational biologists to easily spin up Jupyter notebooks or Rstudio instances with direct access to the counts from nf-core/rnaseq and DESeq2 (as well as all data on the platform).

Create custom dashboards and hand-off to scientists
The dashboards that allow scientists to visualize differentially expressed genes, or discover gene sets and functional annotations, are pre-built examples of a more general dashboarding tool. These existing dashboards can be copied, and used as a base for custom extension, for all types of interesting applications, eg. overlaying biochemical datastreams like qPCR, aggregating many existing experiments.
Install Latch for your Scientific Teams
Latch is a modular and highly programmable data infrastructure designed to orchestrate wet and dry lab teams for deeper + faster biological consensus. For more information about the components of the platform, peruse our documentation:
We work with over 100 biotechs, ranging from fledgling startup to top 20 biopharma, and invest heavily in support and customer obsession. Our bioinformatics and engineering services team would love to partner with you on your next project. Meet our team.
—
Here are some resources that might be helpful self-serving these tools:
Hi Kenny. Nice post. Particularly interested in the sentence "The schemas link this metadata directly to both raw sequencing files and processed results, like gene counts and downstream reports". I know how with latch the metadata can be linked to the raw sequencing data but am unsure how a row can be linked to results, particularly when you could have multiple executions for one sample. Be keen to know more