
The Cloud for Biology
Pre-configured for high-throughput biological data, enabling immediate analysis following best data practices.


Introduction
It’s become a cliche that biotechnology is dealing with an exponential influx of data due to the rise of high-throughput technologies (a prime example being the graph of NGS sequencing costs decreasing shown at the start of every presentation discussing the industry, we’ve been guilty of this ourselves).
To accommodate this data, we are going to need biospecific data infrastructure, but given the nascency of many biological data modalities, the industry has to-date relied on strung together single-point solutions that are underpowered for the task.
Let’s take the computational workflow for one of the most common NGS assays being run today in biology — RNA-seq — as an example to see why this is the case:
Storing and transferring large sequencing files, such as FASTQs, poses challenges. While AWS S3 often serves as a go-to storage solution, it falls short in user-friendliness for scientists. It lacks capabilities for biological visualizations and doesn't provide data/code versioning and traceability, all of which hamper the pace of R&D. Alternative options: Google Cloud, Box, Dropbox, Azure, Finder, Egnyte, and Drive.
Linking raw files with sample metadata is unintuitive. While some companies use a CSV sample sheet because it's user-friendly for biologists, it lacks system integrations, input validation, and API querying capabilities. Others use a Postgres database with a custom frontend, this helps to tackle those challenges but is an overkill in engineering complexity. Other options: Benchling Registry, Excel, Notion, Airtable, Postgres, Quilt, Kaleidoscope, MySQL.
Setting up data pipelines to convert FASTQs into something interpretable, like a Count Matrix, is a labor-intensive task. Many opt for AWS Batch combined with Nexflow, but its extended setup duration can drain resources, and its absence of a user-friendly GUI disempowers scientists. Given sufficient resources, Streamlit becomes a preferred choice for creating a tailored execution launch interface. Other popular tools include Flyte, DAGster, Airflow, NF-tower, SnakeMake, Redun, and Bash.
Configuring computational notebooks and visualizations, to transform a count matrix to interactive gene expression volcano plots is error-prone and repetitive. Many resort to AWS EC2 paired with an iPython notebook, tunneled to the user's laptop. However, onboarding new scientists can be intricate, particularly when installing library dependencies. Alternatives: RStudio, Rshiny, Dash, Jupyter Labs, Colab, Sagemaker, Code Ocean, Watershed, Saturn Cloud.
Thus, a single RNA-Seq experiment showcases the complexities of analyzing vast biological data, which can hinder R&D, affect morale, and inflate costs.
It’s important to remember that these problems are true for single-cell, spatial biology, flow cytometry, and every other high-throughput assay.
Solutions like AWS et al. are hard to configure and demand significant maintenance efforts. All of it to reinvent the same platform as every other data team, while diverting resources away from the company's core scientific differentiators.

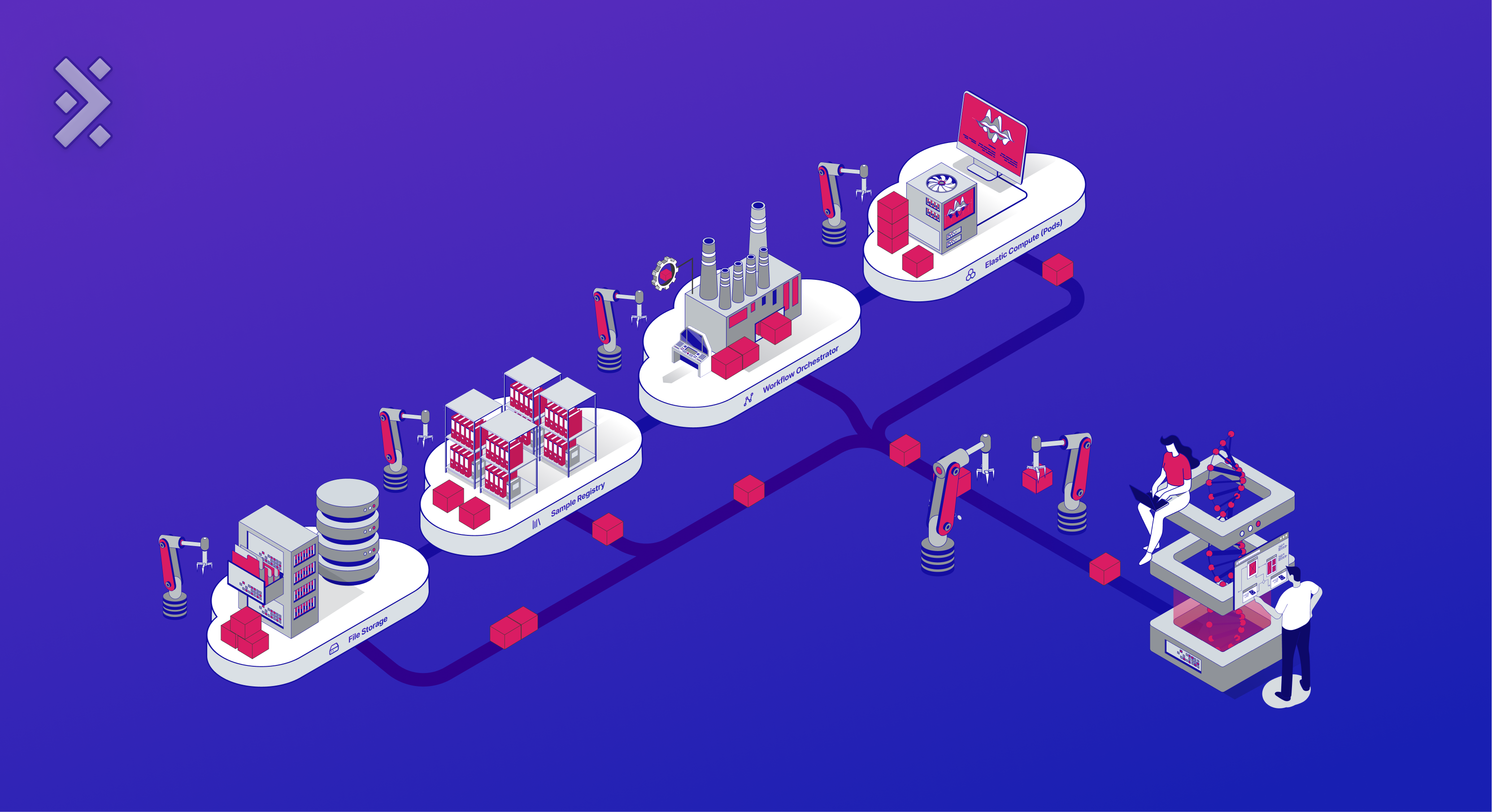
A Cloud for Biology
We propose a specialized cloud, pre-configured to handle biological data, and enabling immediate data analysis. Through continuous collaboration with 50+ biopharma customers across many roles — bioinformaticians, computational biologists, senior scientists, and executives — we've developed a Cloud for Biology.
This system comprises four core cloud components: File Storage, Sample Registry, Workflow Orchestrator, and Elastic Compute. This article delves into these components, showcasing their application in an RNA-Seq experiment and underlining the benefits of a unified Cloud for Biology for biotech data management.

File Storage: A Data Store Accessible to Everyone
The first component of a Cloud for Biology is File Storage. This system holds all data types, from raw FASTQ or BAM files to processed outcomes like PDB and IPYNB files. It should offer biological visualizations, secure access, and robust versioning and traceability that meets FDA reproducibility standards.
It should have a friendly GUI for biologists to explore and an ergonomic SDK and API for developers to query.
In the context of an RNA-Seq experiment, this system becomes crucial. It should effortlessly integrate FASTQ files from sources like Illumina BaseSpace, offer visualization tools like MultiQC, and maintain data integrity with seamless versioning and traceability through every analysis phase (FASTQ -> Count Matrix -> HTML).

Sample Registry: Integrating and Keeping Track of Your Samples
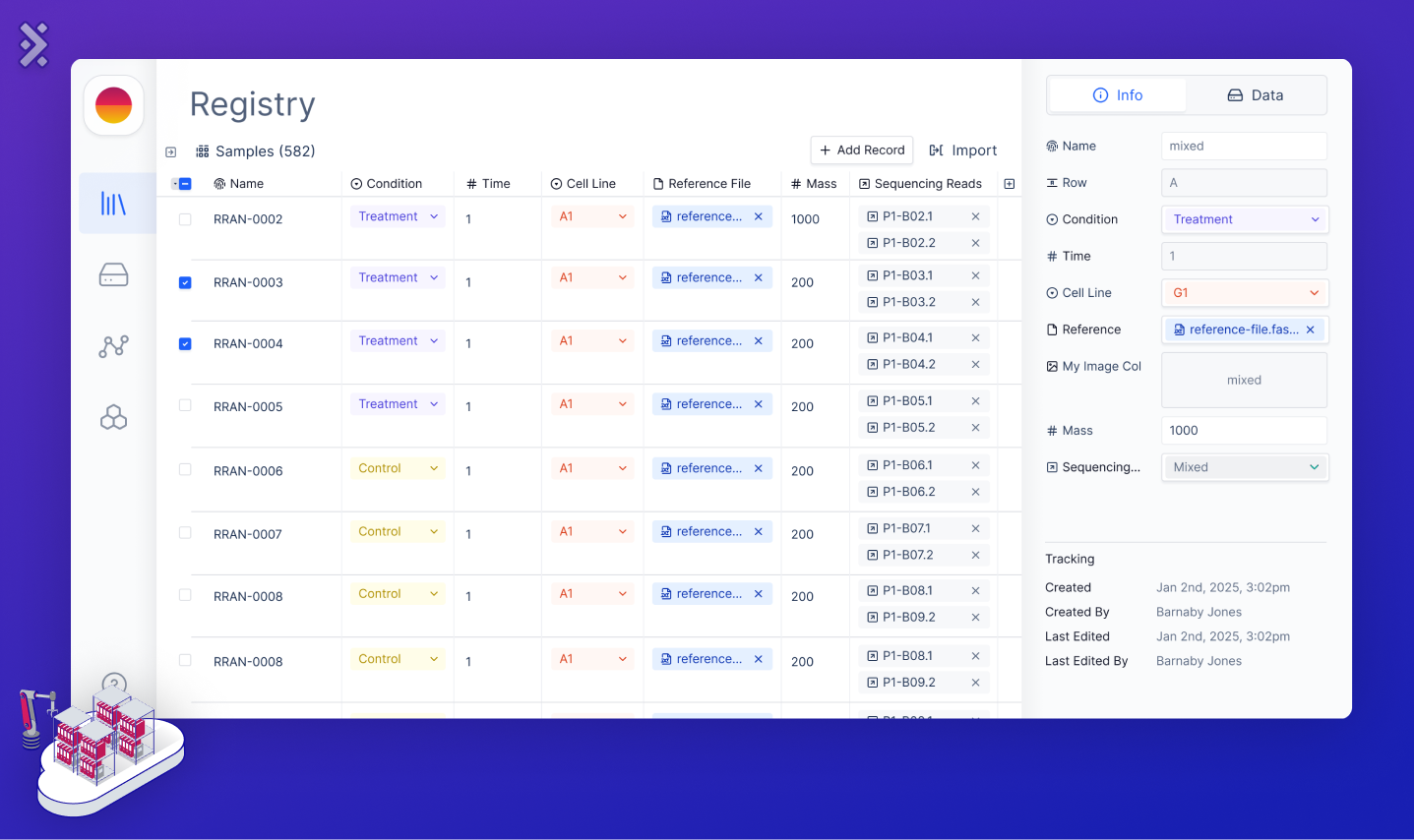
The second component is the Sample Registry, a database for tracking samples and their metadata. This system aids in decision-making, project oversight, and regulatory adherence by consolidating vital metadata. Rather than replacing ELN and LIMS, it links with them, completing an integrated ecosystem for large biological data.
Balancing the needs of developers who prefer relational databases with biologists who prefer Excel is difficult. The Latch Sample Registry accommodates both, ensuring intuitive quality-controlled data entry, efficient search, and dependable access.
This is one area where the tight integrations possible within a cloud platform really shine. Being able to link from file storage directly to Registry, executing a workflow directly from a Registry table, and allowing workflows to read and write directly from the tables.
In the context of RNA-Seq experiments, Registry captures essential experimental metadata, including origins, dates, and conditions (e.g. control vs. treatment). As the biologist fills out the table with their experimental parameters, it should give them instant feedback about any errors/typos that would make the pipeline fail. It should also reference the respective FASTQ file for each sample directly and feed metadata into the RNA-Seq workflows, paving the way to the next component, the Workflow Orchestrator. As such, the Sample Registry is a bridge linking data to analytics, underpinning traceability, reproducibility, and observability — the trifecta of solid scientific research.
Workflow Orchestrator: Scaling and Managing Your Pipelines
The third cornerstone of a Cloud for Biology is the Workflow Orchestrator. More than just initiating data pipelines, it transforms massive data into interpretable insights, merging user-friendly interfaces with intricate logic capabilities.
Crafting a robust in-house orchestrator can be deceptively complex.
The Latch Workflow Orchestrator automatically turns pipelines into no-code interfaces for biologists, offers scalable serverless environments for bio-developers, and streamlines the uploading of new workflows by integrating with popular languages like Nextflow and Snakemake.
In our RNA-Seq example, the Orchestrator processes raw FASTQ files from File Storage, pulls metadata from the Sample Registry, runs the RNA-Seq pipeline, and outputs gene expression matrices. Automating tasks like mapping and differential expression minimizes errors, boosts efficiency, and saves time. By automatically having a user-friendly GUI, it allows self-service by wet lab scientists. It also draws on metadata from the Sample Registry and writes back a new column that links the expression matrix to its respective sample, showcasing the seamless interplay of the system's components.

Elastic Compute (Pods): On-demand computers to run flexible analyses and visualize data.
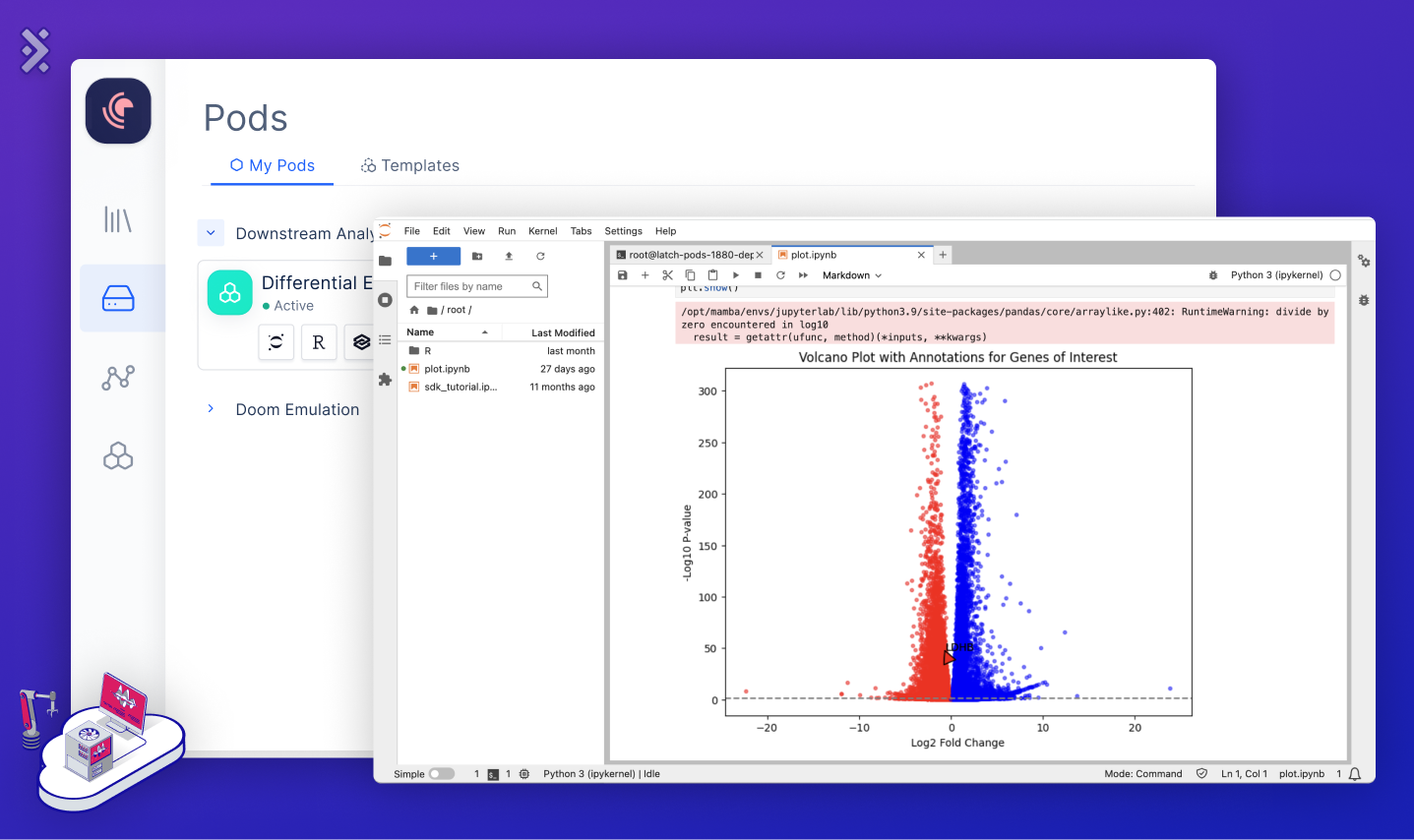
The final piece of a Cloud for Biology is Elastic Compute, or Pods, offering adaptive computational resources for both developers and biologists. Developers should experience an EC2-like environment with SSH access, GPU support, instance resizing, VS Code access, and auto-shutoff. Meanwhile, biologists enjoy user-friendly tools like Jupyter Notebook, RStudio, or dashboards using RShiny and DashPlotly. Integration with the File Storage ensures smooth file access for everyone.
In an RNA-Seq context, Pods shine during the final data analysis. Once gene expression matrices are set, biologists can launch tools like Jupyter or RStudio, run DESeq2, and craft visuals like volcano plots or heatmaps, deepening insights into gene expression variations. Once a final plot is created, we can easily save it to the file storage and link it back to its respective registry sample.
Interfaces and Cloud Hosting
A great Cloud for Biology should support the biologists who prefer to interact with graphical user interfaces (GUI) and the computational biologists/bioinformaticians who might prefer code interfaces (CLI, API, etc.).
It should not need to be set up on top of another cloud computing provider such as AWS, GCP, or Azure. It is the cloud computing provider, and should just work out-of-the-box, every time, every day, anywhere.
Conclusion: The Future of Big Data Management in Data-driven Biotechs
A Cloud for Biology, as we envision it, presents an integrated approach to rapidly transforming raw data into actionable insights following best data practices. With its four core components—File Storage, Sample Registry, Workflow Orchestrator, and Elastic Compute—a cloud for biology effectively bridges the gap between the biologists, who want to interact with a GUI, and the developers, who want control and flexibility over their platform. In doing so, it accelerates the speed at which biotech companies can handle, analyze, and interpret big data. Streamlining operations, breaking down data/code silos, fostering cross-disciplinary collaboration, and accelerating the pace of scientific discovery.
—
If you are currently setting up a cloud for biology, whether you choose to use Latch or not, we would love to hear about your experience and help however we can. We love feedback, enjoy engaging with the community, and we use both to keep improving. Please reach out to alfredo@latch.bio.
Latch is free to sign up for and gives $100 of free credits to every user. Anyone can sign up at console.latch.bio.
Thanks to Aidan Abdulali, Hannah Le, Kenny Workman, Tess van Stekelenburg, Kyle Giffin, and Niko McCarty for suggestions and for reading drafts of this post.