
Virtual Screening for New Antibiotics: A Machine Learning Approach to LpxC Inhibition
A computational search through 250,000 compounds for tomorrow's antibiotics against gram-negative bacteria

With antibiotic resistance threatening millions of lives and pharmaceutical companies abandoning antibiotic development, machine learning-powered virtual screening of chemical databases offers a promising approach to discover urgently needed antibiotics. We conducted our own virtual screening campaign, analyzing 250,000 compounds to search for potential antibiotics that target LpxC, an essential enzyme in gram-negative bacteria.
In this blog, we cover:
Building a compound library
Predicting clinical toxicity, bioavailability and solubility properties of library compounds with ADMET-AI
Using GNINA's ML-powered molecular docking to predict binding affinity to this bacterial enzyme
The Halicin Blueprint

The story of halicin stands out as a remarkable chapter in modern drug discovery. Originally developed in 2009 at the Burnham Institute for Medical Research as a potential treatment for diabetes, no one could have predicted that it would make a remarkable comeback as an effective antibiotic against resistant bacterial strains. In 2020, MIT researchers used a machine learning model to screen the Broad Institute's Drug Repurposing Hub of 6,000 compounds, identifying halicin as having promising antibacterial properties [1]. This rediscovery highlights an exciting possibility: our existing chemical libraries contain hidden antibiotic treasures that are waiting to be discovered with virtual screening.
Where have all the antibiotics gone?
Historically, antibiotics were discovered primarily by analyzing soil microorganisms for compounds that inhibited bacterial growth [2]. Following the discoveries of penicillin, tyrocidine and numerous reports of the production of antimicrobial compounds by microorganisms, Selman Waksman systematically studied microbial compounds that kill other microbes in the late 1930s. Often called the “Father of Antibiotics”, he pioneered modern antibiotic discovery and his research launched the field's Golden Age (1940s-1960s), during which many antibiotics still used today were discovered. He also notably coined the term “antibiotic” as a compound made by a microbe to destroy other microbes [3]. The search for novel antibiotics since this Golden Age has stalled on two fronts: researchers keep rediscovering known molecules in natural product screening (dereplication problem), while attempts to modify existing antibiotic structures rarely succeed despite numerous possible chemical variations [4,5].

Despite the importance of antibiotics in modern medicine, they are no longer lucrative to develop, with the cost of development (~$1B USD) far exceeding their earning potential (~$100M USD annually) [6]. Novartis, AstraZeneca, Roche, Bristol-Myers Squibb and Eli Lilly have all announced that they will no longer be developing antibiotics [7]. With few new antibiotics in the pipeline and bacteria only getting more resistant, antimicrobial resistance is projected to claim more lives than cancer by 2050 [8].
The potential of ML-driven virtual screening
Public databases contain billions of synthesizable compounds, making manual screening of potential antibiotics impossible. Machine learning solves this by analyzing molecular structures for patterns linked to antibiotic activity at massive scale. The Collins and Barzilay Labs at MIT proved this approach in 2020: their deep learning model, trained to recognize antibiotic-like molecular structures, screened 6,000 compounds from the Broad’s Drug Repurposing Hub and identified halicin — then simply a diabetes drug with unexpected antibacterial potential [2].
In short, the wet lab validation of halicin that followed proved remarkable. Over 30 days, the bacteria E. coli showed no resistance to halicin. In contrast, E. coli developed resistance to the traditional antibiotic ciprofloxacin within just 1-3 days [9]. Halicin also showed broad-spectrum antibiotic activity in mice.

An interesting side note: halicin was named after the fictional artificial intelligence HAL from 2001: A Space Odyssey.
The anatomy of a virtual screening campaign
Inspired by the rediscovery of halicin, we decided to run our own virtual screening campaign to identify potential antibiotic compounds from existing public databases. There are a lot of new tools being published to narrow down candidates, so we took this as an opportunity to put them to the test.
Instead of predicting antibiotic activity for a library of compounds (the approach taken by the MIT research teams), we chose a more generalizable approach — screening ligands for their predicted binding affinity to a therapeutically relevant target.
This post outlines the following steps:
Downloading 250k compounds compounds from CartBlanche22 (in SMILES format)
Predicting ADMET properties with ADMET-AI to arrive at 630 candidate compounds
Converting the SMILES representations of the compounds to 3D structures stored in .sdf files
Using GNINA to predict docking affinity values for these compounds against our selected target
Disclaimer: We are not claiming to have done groundbreaking science here. We find this problem interesting and are merely demonstrating an example of how you can use publicly available tools to screen through chemical databases.
Target selection: LpxC
We chose the bacterial enzyme LpxC as our bacterial target, also known by the lengthy alias of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase. The goal of designing an inhibitor for LpxC is to bind to and block the enzyme's activity.
LpxC is essential for lipid A biosynthesis in gram-negative bacteria and represents a promising antimicrobial target due to its absence of human homologs and critical role in bacterial survival. Despite its attractive properties - including cytoplasmic location, well-characterized binding pocket, and multiple available crystal structures - development of clinically viable inhibitors has been challenging. While Merck scientists first described LpxC inhibitors in 1996, only two candidates (ACHN-975 and RC-01) have reached clinical trials, both terminated in Phase I due to safety concerns. The primary obstacles aren't antimicrobial efficacy but rather toxicity issues. The target remains compelling because of its necessity for dangerous pathogens (E. coli, P. aeruginosa, K. pneumoniae), low mutation rate, absence of alternative pathways, and lack of known natural resistance mechanisms [10].
1 - Building a chemical library
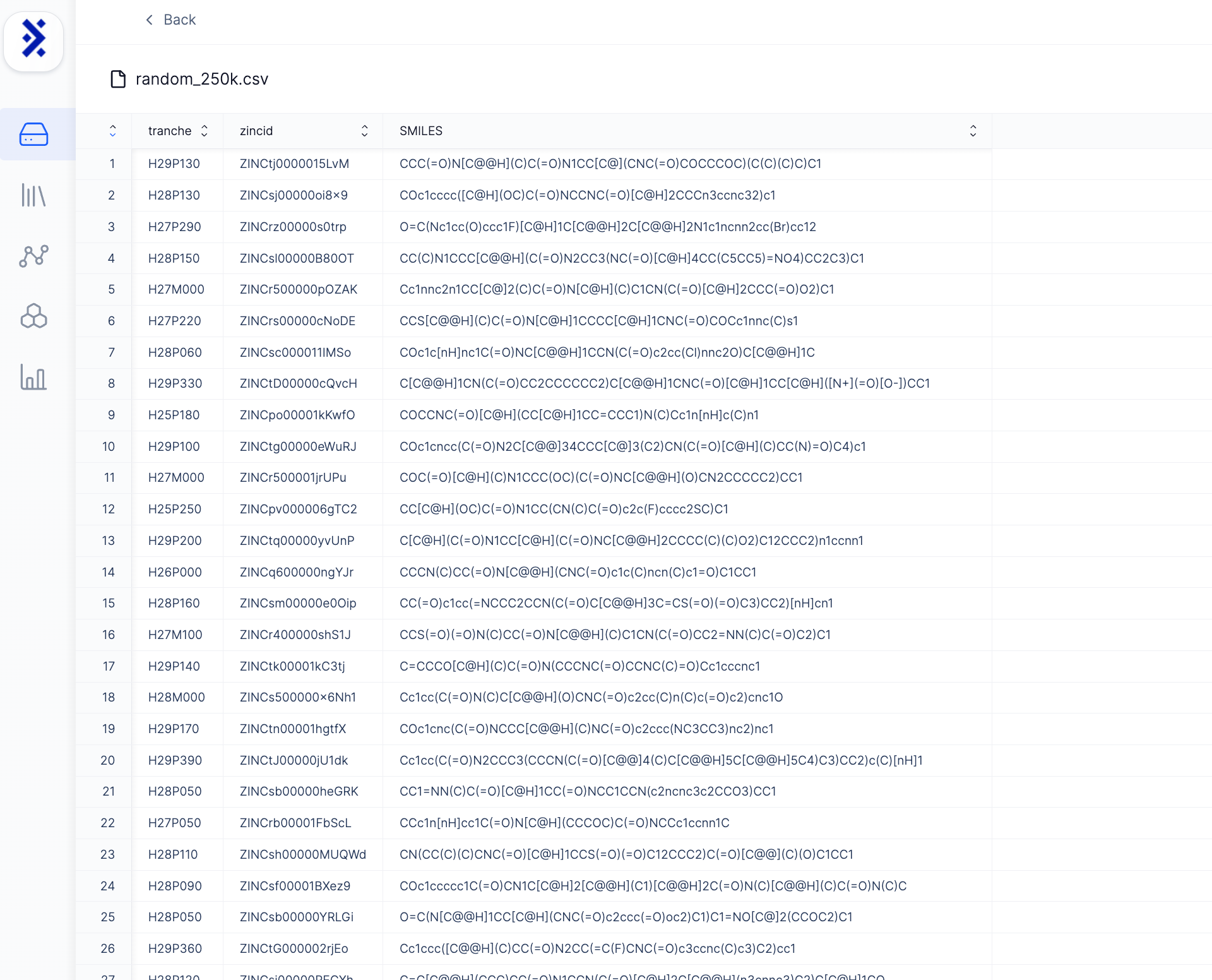
The first step in any virtual screening campaign is acquiring a diverse set of chemical compounds to evaluate. We utilized CartBlanche22, an awesome resource compiled by UCSF’s Irwin and Shoichet Labs that provides free access to purchasable, drug-like compounds. It contains ~54.9 billion molecules in 2D and ~5.9 billion in 3D [11]. We randomly downloaded 250,000 compounds in SMILES format to cast a wide initial net (still a very small subset of CartBlanche22's full library).
We also downloaded LpxC from the RCSB PDB database: 3p3e.pdb
2 - Predicting ADME properties
Starting with a CSV of the 250k SMILES strings, we used ADMET-AI to predict key pharmacological properties. Developed through a collaboration between Greenstone Biosciences and Prof. James Zou's lab at Stanford, ADMET-AI uses machine learning models trained on experimental data to predict absorption, distribution, metabolism, excretion, and toxicity properties [12].
This screening step is crucial for two reasons:
Computational efficiency - no need to dock compounds that would make poor drugs
Focus on viable candidates - particularly important given LpxC inhibitors' history of toxicity issues in clinical trials
ADMET-AI also compares each compound's predictions to predictions for approved drugs. The team created a reference set of 2,579 drugs from the DrugBank that have obtained regulatory approval and applied ADMET-AI to make ADMET predictions for all of these molecules. For every compound run through ADMET-AI, it computes the percentile of the ADMET predictions with respect to these 2,579 reference approved drugs. For example, if a molecule receives a bioavailability percentile of 90%, it means that the molecule is more bioavailable than 90% of approved drugs in the reference set. They have an awesome blog post that shares more [12].
We used the ADMET-AI workflow on Latch to predict these properties for the 250k compounds, which took just 48 minutes!
We then wanted to keep compounds that had percentiles above 50% for the following properties, with respect to the reference DrugBank set:
Bioavailability:
Oral bioavailability percentile. Higher percentile indicates better oral bioavailability.
Solubility:
Aqueous solubility percentile. Higher percentile indicates better solubility.
BBB:
Blood-Brain Barrier permeability percentile. Higher percentile (after inversion) means less BBB penetration, which is better to avoid neurotoxicity.
hERG:
hERG channel inhibition percentile. Higher percentile (after inversion) means lower cardiotoxicity risk compared to approved drugs.
Clintox:
Clinical trial toxicity percentile. Higher percentile (after inversion) means lower toxicity compared to approved drugs.
This stringent filtering step reduced the compound set from 250,000 to 630 candidates. You can see here a plot of the average score across these categories for these 630 candidates, some of which were >80.0:
3 - Adding another dimension
For the tools we used at later stages of this workflow, it was necessary to have a 3D representation of the SMILES strings. For each compound, we used OpenBabel to convert the SMILES strings to 3D .sdf files. Here’s what an example compound looks like:
4 - Predicting affinity with GNINA
Next, we decided to use GNINA to dock these compounds to LpxC. GNINA is a molecular docking program forked from AutoDock Vina with integrated support for scoring and optimizing ligands using convolutional neural networks [14,15]. It runs in 1-2 minutes per compound on modern GPUs — making it ideal for docking large libraries of compounds.

We used it to dock the compounds to LpxC and had scores for 630 compounds in a few minutes. We performed whole protein docking, meaning that the autobox_ligand parameter was set to the PDB file of LpxC.
As another resource, this deck dives further into the details of running GNINA [16].
For one of the ligands, you can see the different poses predicted by GNINA, each of which has associated metrics:
For every compound, GNINA outputs a log file with scores for different poses using four key metrics:
The traditional affinity score (measured in kcal/mol) predicts binding energy, where more negative values indicate stronger binding - scores below -8.0 kcal/mol suggest strong interactions.
The intramolecular energy score reveals internal strain in the molecule, ideally staying at or below 0 kcal/mol to indicate a stable conformation.
These traditional metrics are complemented by two ML-driven scores:
The CNN pose score (0-1) reflects the models confidence in the predicted binding position, with values above 0.5 suggesting reliable predictions.
The CNN affinity score (0-10) provides a machine learning estimate of binding strength, where higher values generally indicate stronger interactions.
Filtering on some of these columns, we can see the most promising compounds at the top of the table:
An example “top“ compound: ZINCn500001tBjEc
Pose 1 of compound ZINCn500001tBjEc seems like one of the the most promising compound, given its metrics:
affinity: -7.69
intramol: -0.66
cnn_pose_score: 0.7689
cnn_affinity: 6.052
For reference, this is its associated SMILES string: CNC(=O)CN=c1[nH]c(=NCc2cnnn2C)[nH]c2c1CCC2
What’s next?
In just a few hours, it was possible to screen thousands of compounds by leveraging this new wave of machine learning tools. While virtual screening can identify promising candidates, it's just the first step in a long journey toward discovering new antibiotics. From here, you could screen top candidates with complementary tools, perform molecular dynamics simulations to better understand binding stability, or analyze specific protein-ligand interactions in the binding pocket.
The identification of compounds like ZINCn500001tBjEc, illustrates how virtual screening could uncover new chemical scaffolds for antibiotic development. While extensive lab validation would be needed, using this approach to prioritize which compounds to test experimentally saves millions in development costs. As antibiotic resistance continues to grow as a global health threat, these efficient screening methods are crucial for building our medical arsenal against bacterial infections.
—
All tools used in this workflow (and more, like DiffDock) are publicly available on latch.bio, meaning you can choose your own target and replicate this flow. Feel free to send an email to bronte@latch.bio or book a demo if you want to learn more!
Thank you to Tahir D'Mello, who built the infrastructure supporting these tools, and Kenny Workman, who provided thoughtful review. Their dedication to democratizing cool scientific tools continues to inspire.
References
https://betterworld.mit.edu/artificial-intelligence-yields-new-antibiotic/
https://news.mit.edu/2020/artificial-intelligence-identifies-new-antibiotic-0220
Irwin, JCIM, 2022, submitted. https://doi.org/10.26434/chemrxiv-2022-82czl
Swanson, Kyle, et al. "ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries." Bioinformatics 40.7 (2024): btae416.
McNutt, Andrew T., et al. "GNINA 1.0: molecular docking with deep learning." Journal of cheminformatics 13.1 (2021): 43.
This is so cool Brontë! Curious what some of the standard antibiotics that we know work would score on those dimensions you show at the end!
Also, should/can that winning molecule be ordered and tested on some E. coli asap..? 😁