
A New Spatial Epigenomics Data Portal
New datasets available to explore the spatial epigenetic landscape // created by AtlasXOmics' DBit-seq technology

Last week we released 25 million spatial transcriptomic profiles on Latch. These datasets allow scientists to ask questions at scale about how genes are expressed across tissues, diseases, and organisms.
Yet understanding the complete state of a cell requires more than measuring RNA. Gene expression is the outcome of a complex regulatory program, influenced by chromatin accessibility, histone modifications, and DNA methylation. Capturing these modalities in their native spatial context is essential for building a more complete atlas of biology.
Today, we are releasing a new data portal with AtlasXomics dedicated to spatial epigenomics. The portal includes datasets from four publications, spanning diseases such as Alzheimer’s and gastric adenocarcinoma, along with studies of heart and brain tissue development.
These resources bring a new layer of information into focus, revealing not only which genes are expressed, but also how the regulatory architecture of the genome shapes that expression in space.
Visit the portal and study these datasets here: https://atlasxomics.latch.bio/data-portal
How does it work?
At the heart of these datasets is a technique called Deterministic Barcoding in Tissue sequencing (DBiT-seq), pioneered by AtlasXOmics. DBiT-seq uses microfluidic chips to flow barcoded oligonucleotides across tissue sections. Each intersection of these flows marks a defined spatial coordinate, which can then be linked to sequencing reads.
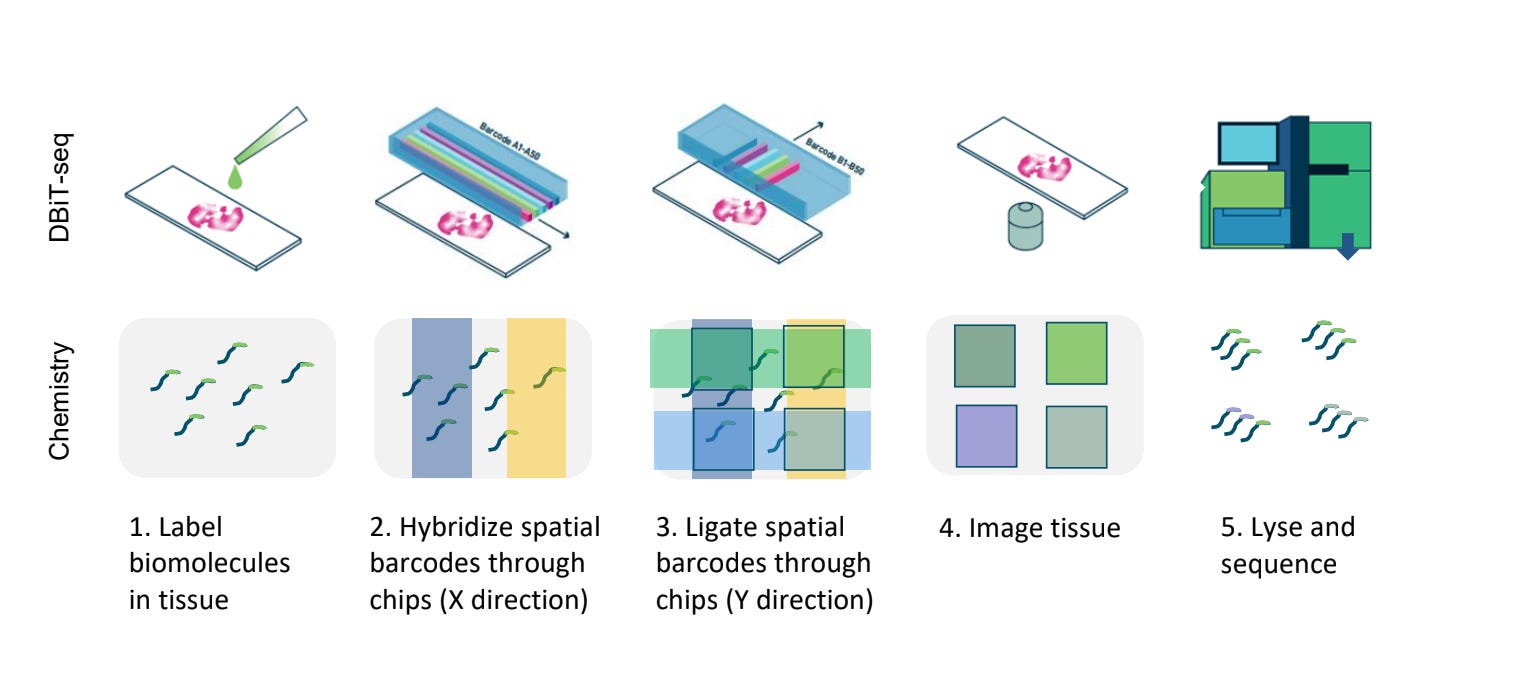
The result is a generalizable platform that can add spatial context to a variety of assays. When combined with ATAC-seq, for example, DBiT-seq produces spatially resolved maps of chromatin accessibility. Similarly, paired with CUT&Tag, it reveals the distribution of histone modifications such as H3K27ac or H3K4me3 across tissue.
What does the data tell us?
To understand what insights are revealed through DBiT-seq, let’s walk through an example dataset of how spatial ATAC-seq helps us better understand tumor heterogeneity and chromatin dysregulation in two stomach carcinoma patient samples.
Tumor heterogeneity remains a formidable challenge in cancer research and treatment, manifesting from inter-patient variability to intra-tumor clonal diversity. This heterogeneity complicates understanding of disease progression and therapeutic response. At the molecular level, chromatin dysregulation is a key driver of heterogeneity: aberrant chromatin states influence gene expression, cell identity, and tumor microenvironment (TME) interactions, all of which contribute to resistance pathways and metastasis.
Spatial ATAC-seq combines chromatin accessibility profiling with spatial resolution, allowing researchers to identify regulatory elements active in specific regions of a tissue. By coupling high-throughput sequencing with spatial barcoding approaches, the AtlasXomics platform enables:
10µm resolution: Identification of epigenetic states at the single-cell level.
Spatial context: Mapping chromatin landscapes to morphological features.
Comprehensive analysis: Integration of gene activity scores, motif enrichment, and peak accessibility with spatial representation.
These capabilities distinguish spatial ATAC-seq from bulk or single-cell ATAC-seq by preserving the physical location of cells, enabling deeper insights into tumor biology.
Mapping cells in TME

This dataset demonstrates how spatial ATAC-seq can disentangle cell state differences between tumor and surrounding normal tissue. By quantifying chromatin accessibility and inferring gene activity scores, it becomes possible to delineate distinct biological compartments and cell types, such as immune, muscle, and tumor-specific populations.
The UMAP projection below highlights major clusters including tumor, mucosa, muscle, and mixed tumor-immune states. When integrated with spatial mapping, these clusters align with morphological regions of the tissue, revealing how chromatin accessibility defines functional neighborhoods within the tumor microenvironment.
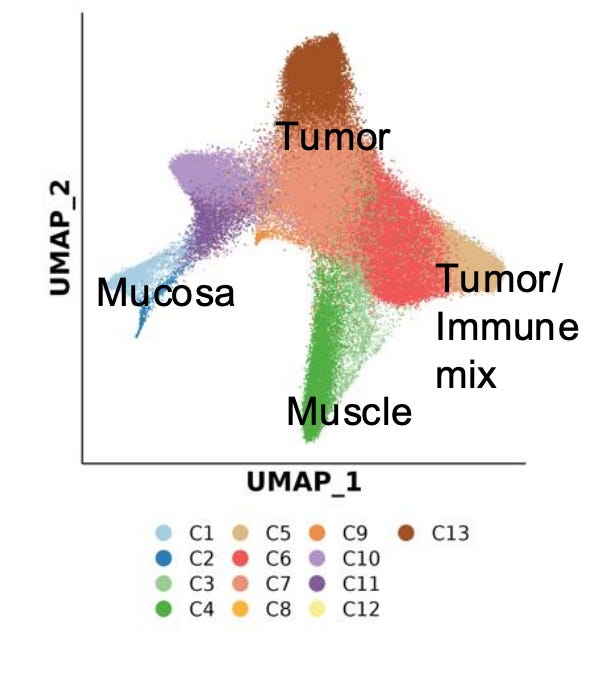
Such spatial alignment is essential for understanding how local chromatin states shape cellular interactions, influence immune infiltration, and drive tumor heterogeneity.
Deciphering Spatial Chromatin Dysregulation in TME
Chromatin accessibility reflects the interactions of DNA and transcription factors, and therefore a lens through which one can study underlying cell states in a tumor that lead to specific gene expression programs.
Increased accessibility may allow cancer cells to activate survival programs, upregulating anti-apoptotic genes (e.g., BCL2), stress response regulators, and genes promoting immune suppression (e.g., PD-L1, IDO1/IDO2 ) , enabling adaptation to therapy and ultimately, resistance. Resistant clones can exhibit unique chromatin landscapes with enriched accessibility at loci controlling drug efflux, transcription factors (e.g., MYC), and epigenetic modifiers.
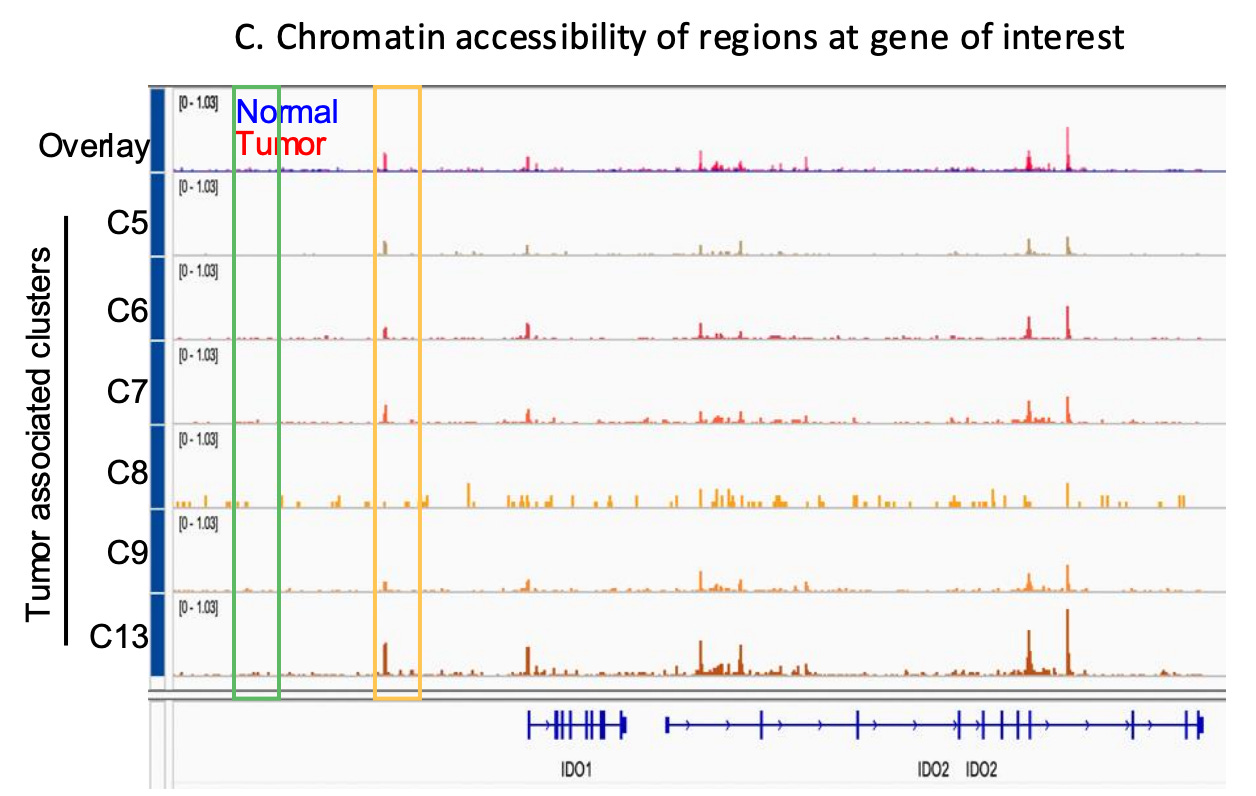
Targeting chromatin regulators, such as histone deacetylases (HDACs) or bromodomain and extraterminal domain (BET) proteins, holds promise as a therapeutic strategy to counter these adaptations and re-sensitize tumors to treatment.
Differential Gene Accessibility Defines Distinct Tumor Compartments
Differential gene accessibility can reveal how distinct tumor compartments are functionally organized. By mapping the spatial distribution of highly differentially accessible genes, researchers can identify how cellular neighborhoods are poised to take on specialized roles of specific TME niches,such as immune hot vs. cold, invasive, or hypoxic environments.
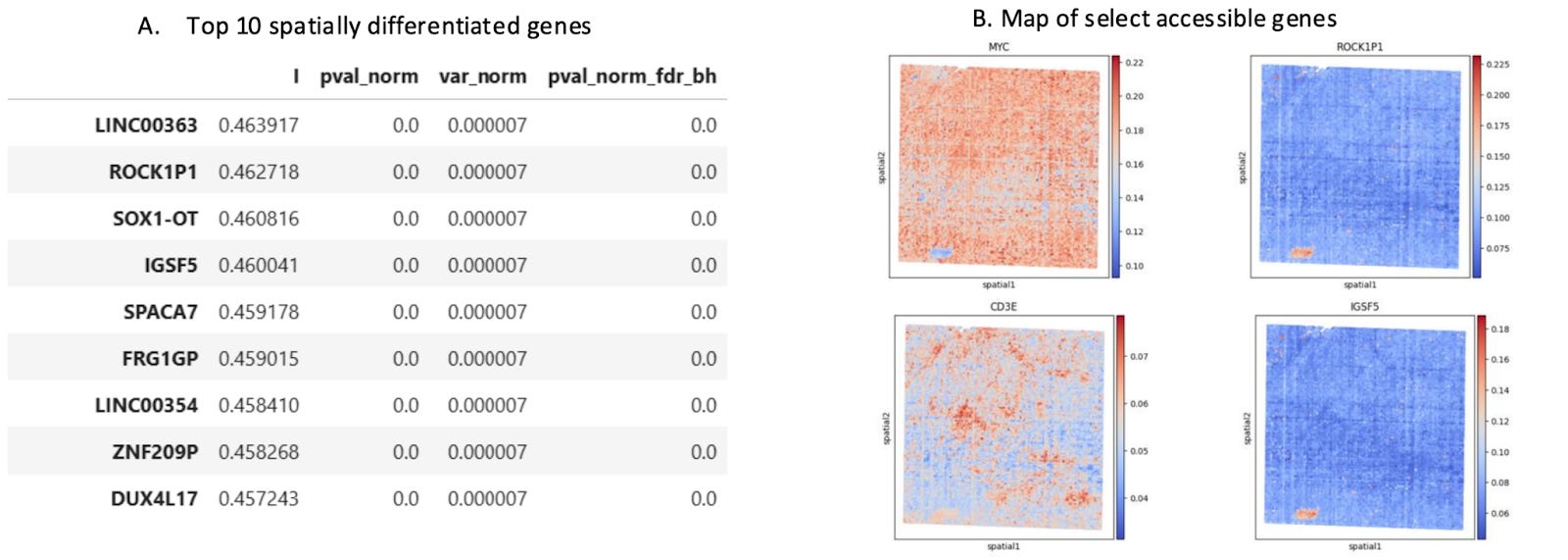
Looking for Drivers of Disease
Examining motif enrichment in tumors compared to matching normal tissue reveals the regulatory elements driving cancer progression. Differential motif analysis reveals transcription factors and expression programs uniquely activated in tumors, shedding light on the epigenetic reprogramming that underpins oncogenesis. This approach can identify key cancer drivers, such as transcription factors supporting unchecked cell growth or immune evasion, and distinguish regulatory pathways associated with therapy resistance.
Additionally, comparing motif enrichment between tumor and normal tissues helps delineate how chromatin accessibility shifts contribute to tumor heterogeneity, highlighting the diverse adaptations of cancer cells to their microenvironment.
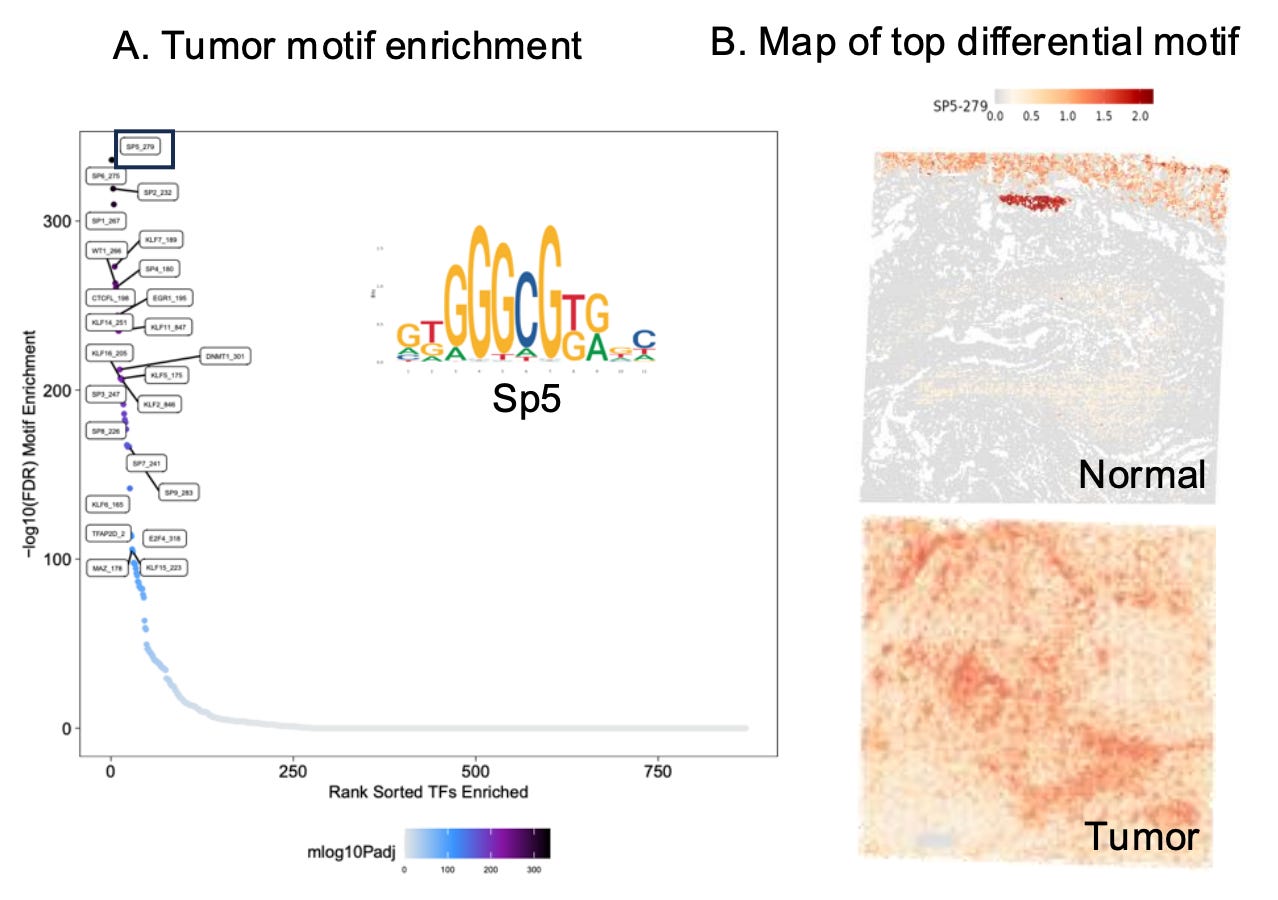
By pinpointing these unique regulatory elements, researchers can uncover novel biomarkers for early detection and identify region-specific therapeutic vulnerabilities.
Explore yourself
Spatial epigenomics presents a powerful framework for linking chromatin states to the physical architecture of tissues. By layering accessibility, histone modifications, and transcriptional readouts onto spatial coordinates, these datasets reveal how regulatory programs shape disease progression, tissue development, and therapeutic response.
Visit atlasxomics.latch.bio/data-portal to visualize and explore yourself.
Contact colinn@atlasxomics.com to learn how you can perform single-cell spatial CUT&TAG, or ATAC-seq in your experiments.
If you are a kit or solution provider interested in setting up your own portal and bundle it with analysis packages, contact alfredo@latch.bio