
Engineering AAVs with Evo and AlphaFold
A modern data stack for machine learning guided drug development

Drug discovery, and much of bioengineering, will progress faster if we are able to engineer molecules to do what we want. Powerful machine learning tools are emerging to aid the development of these biomolecules, often in tight feedback loops with wet lab experiments, that require coordination between wet and dry lab teams and complex software infrastructure.

It is sometimes unclear how to use these tools in practice or what the components of an end to end software system look like in the context of real biology and problems biotechs face in industry. Here we trace the design, build and testing of viable AAV capsids on a single data platform, LatchBio, highlighting design principles and addressing problems coordinating diverse scientific teams at each step:
Finetuning the genomic language model Evo and generating initial designs
Storing molecular libraries in a database accessible to scientists
Screening candidates in-silico with batched AlphaFold
Processing sequencing data from functional validation assays
Synthesizing and visualizing results

We are parameterizing the system with components useful for engineering AAV capsids to ground the discussion in realistic and perhaps familiar biology, but the design principles generalize to machine learning guided workflows for most biologics. The data and components of the loop, including access to Evo and AlphaFold, are linked throughout and available for public exploration and use:
Table of contents
Centralizing molecular libraries with metadata for diverse scientific teams
Closing the loop - drawing meaningful biological conclusions
Machine learning guided engineering loops
Because molecules, and the biological systems they interact with, are very complex, human minds are bad at designing them rationally. We instead use screening techniques to allow us to build and test many thousands of molecular variants simultaneously and select promising designs based on their function in an end-to-end system, like a cell or mouse model. This high-throughput experimental data can be fed into statistical models that suggest new designs, forming a “design-build-test loop” that has become the bedrock of modern bioengineering.
It is becoming clear that the cheaper and faster we can run these loops, the closer we move to reducing biology to a true engineering discipline. As the wet lab constituents - automation, microfluidics, synthesis and sequencing - continue to improve, and more industrial teams invest in the infrastructure and tacit knowledge needed to pool and screen larger libraries of designs, the limiting step of the loop is no longer a single experiment.
Great inefficiencies exist at the junctures between steps, where different wet and dry lab teams must access and handoff data, but currently struggle due to different technical languages and fragmented software. The computational steps are also taking longer and becoming more expensive. State of the art language models for library generation and synthesis can take days and thousands of dollars to train. Bioinformatics workflows to process sequencing data from validation experiments have a similar cost profile.
These are both software problems. Experimental biology, in contrast, suffers from strong physical constraints and despite continued advances in assay throughput and speed, it is hard to make cells grow faster. But the resource waste from poor cross team communication and inefficient or broken computational steps can be solved by engineering a better data platform. They should certainly not be rate limiting in a well constructed loop.
AAVs make a good case study
The focus of this walkthrough is engineering AAVs, viruses widely used by the industry as delivery systems for therapeutic payloads, like gene editing tools. Biotechs (eg. Dyno Therapeutics, 4D Molecular Therapeutics, Capsida Biotherapeutics) are interested in engineering the capsid proteins to target specific tissues or overcome immunity. When the capsid proteins are represented as libraries of sequences, many of the techniques and problems in engineering these libraries are agnostic to the molecules they encode and generalize to other biologics, like antibodies or ASOs.
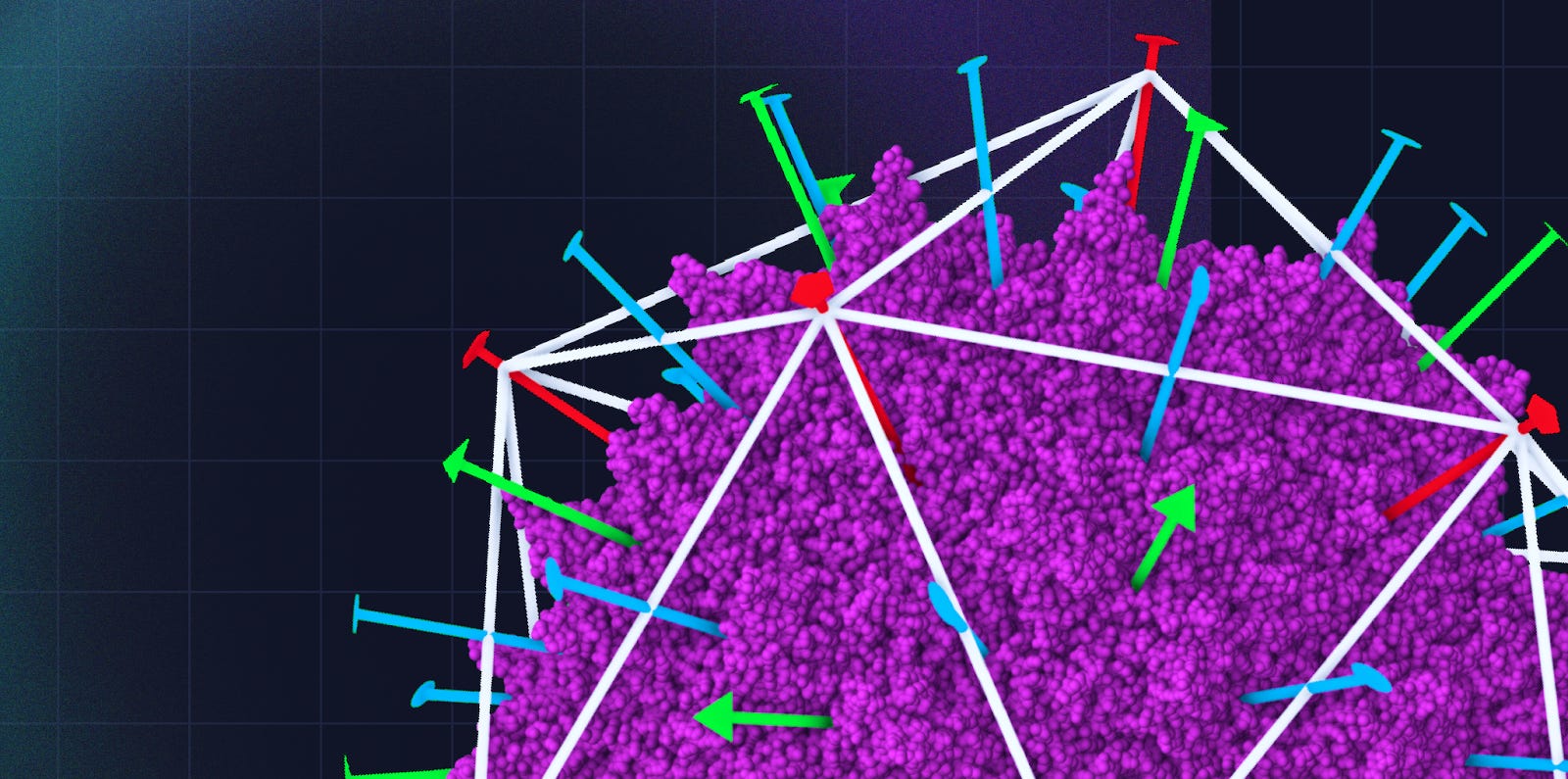
(Figure 4b from Ogden et. al [2])
It is also easy to see how the rational design of these capsids is difficult. The vast majority of changes produce “unviable” capsids that do not assemble or package genetic cargo, and viability alone is only a necessary but not sufficient condition of an engineered virus. Most gain of function variants, especially encoding properties like immune escape, demand sequences that are very different from natural serotypes and require multiple concurrent mutations in protein regions that have a complex geometry, in our example, looping in and out of an “exposed” and “buried” state within the capsid [1]. The complexity of the problem and size of the design space makes not only rational design, but even random mutagenesis, impractical and requires statistical approaches, like machine learning, to learn associations between capsid function and patterns in sequence space.

(Figure from Bryant et. al [1])
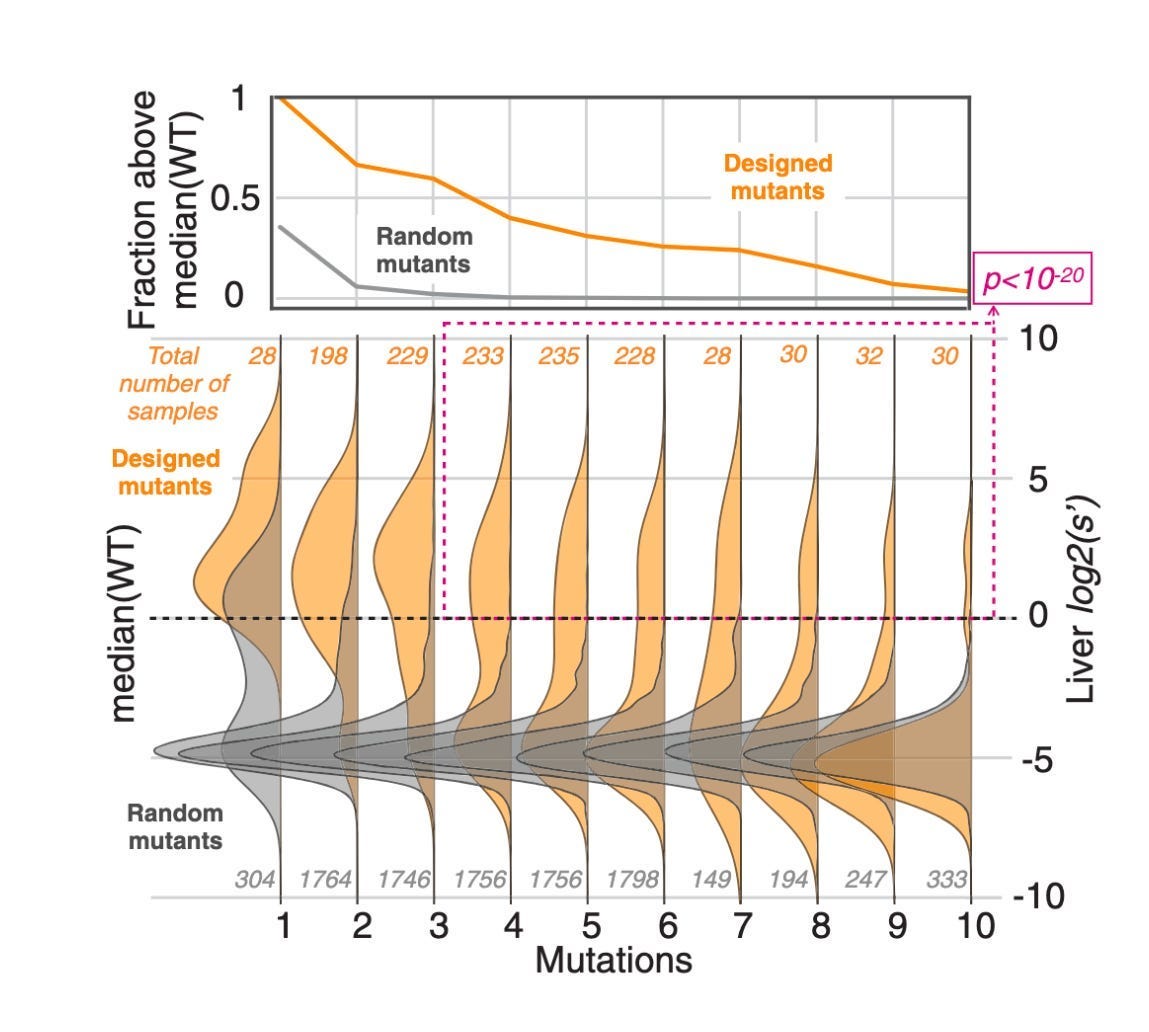
The goal of our engineering loop is to produce viable variants of a small subsequence (positions 561–588) of the AAV2 VP1 protein (highlighted above), that encompasses buried and surface regions and is strongly predictive of function. An initial dataset of these subsequences, along with viability data (the observed counts of each design after transfected into HEK cells), will be used to finetune Evo and is repurposed from Bryant et. al [1], who used it originally to evaluate a collection of statistical methods towards predicting viability. Any results (eg. ability of Evo to generate viable AAVs in practice) are mock and the focus of this work is on the software infrastructure and realistic use of the system by teams in industrial biotech.
Finetuning Evo with in-house datasets
The first component of our engineering loop is a statistical model that learns associations between AAV capsid viability and DNA sequences. This component both generates the initial library and updates its associations with experimental data to generate new designs.
While there are many capable methods in machine learning, we chose to use Evo [3], a large language model with 7B parameters developed by the Arc Institute, that learns complex interactions between genetic patterns without explicit biological modeling. By training on a large corpus of prokaryotic sequencing data, it has demonstrated predictive ability across diverse biological tasks, such the effect of mutation on protein fitness or the expression of a gene from an upstream regulatory sequence.
Because it is a generative model, it can be used to construct sequence libraries encoding arbitrary molecular designs and can also be finetuned with custom datasets to learn the disease biology or molecular properties specific to a drug program, such as viability of AAV capsids.
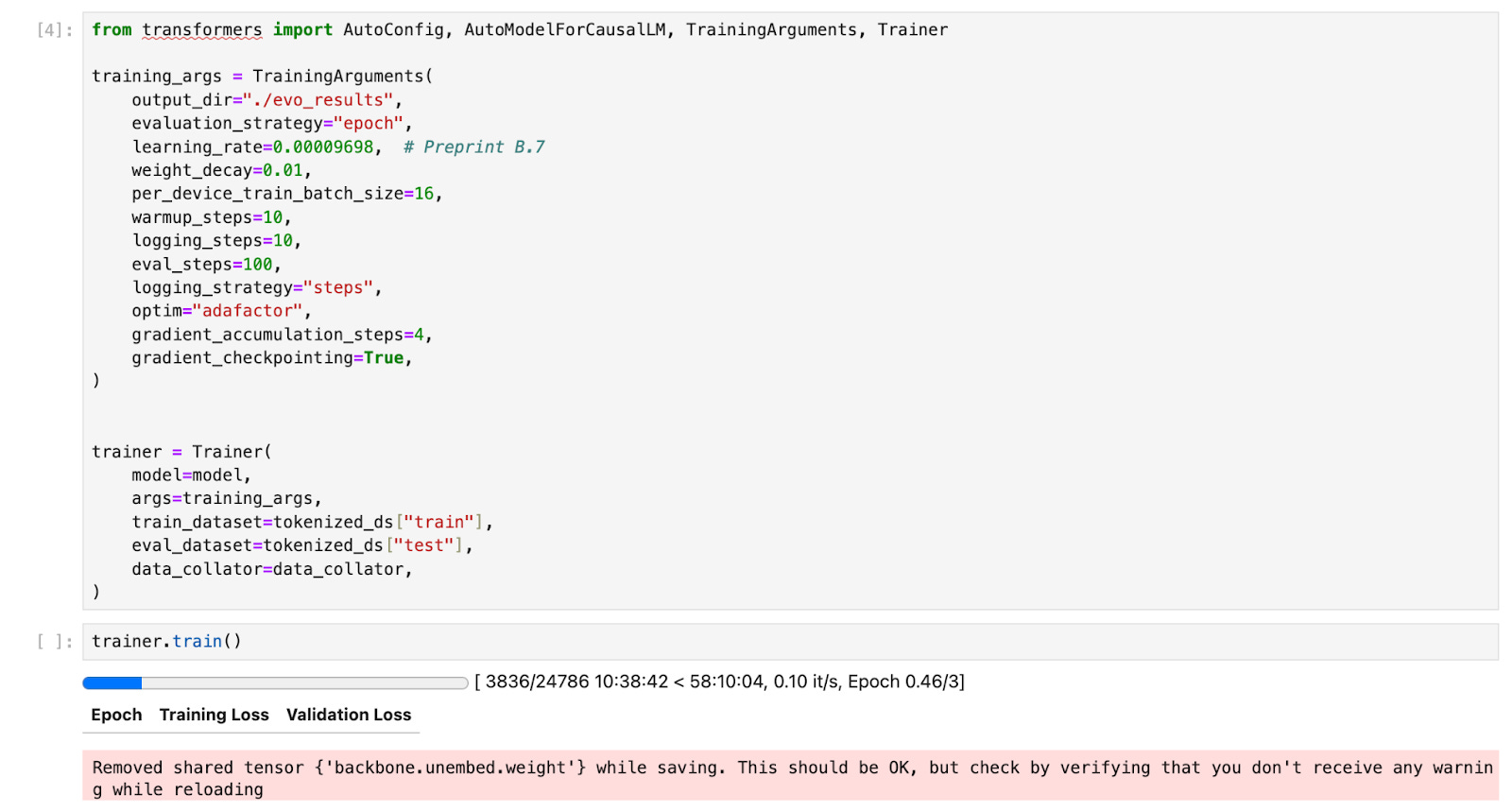
(Finetuning Evo [3] using parameters from Arc’s preprint with Bryant et al’s [1] AAV dataset in a Latch Pod. Notebook boilerplate available in the linked pod template.)
Using Evo requires provisioning GPUs, managing device drivers and installing machine learning libraries from a fast moving and volatile dependency ecosystem. Familiarity with the model API and training libraries to perform tasks like sequence prediction and fine tuning is also required.
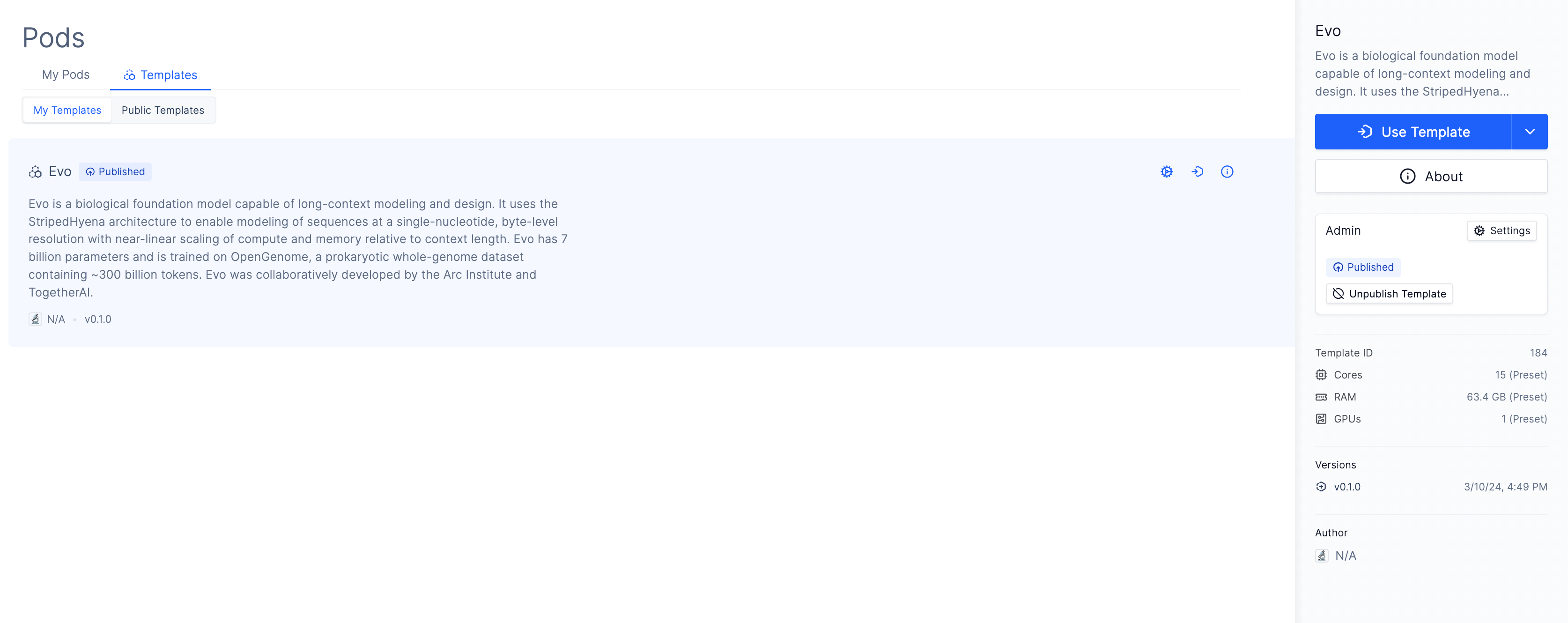
Access to environments with the necessary computing resources and dependencies preconfigured, along with boilerplate code needed for common tasks, reduces the time needed to use Evo. We use an Evo Pod Template to solve these problems.

Centralizing molecular libraries with metadata for diverse scientific teams
After libraries are generated with Evo, the sequences for each design need to be stored in a central database in structured schemas that capture important metadata. Experimental information about the library, such as the iteration round, drug program, intended cell model, as well as statistics from Evo, like sequence entropy, are stored.
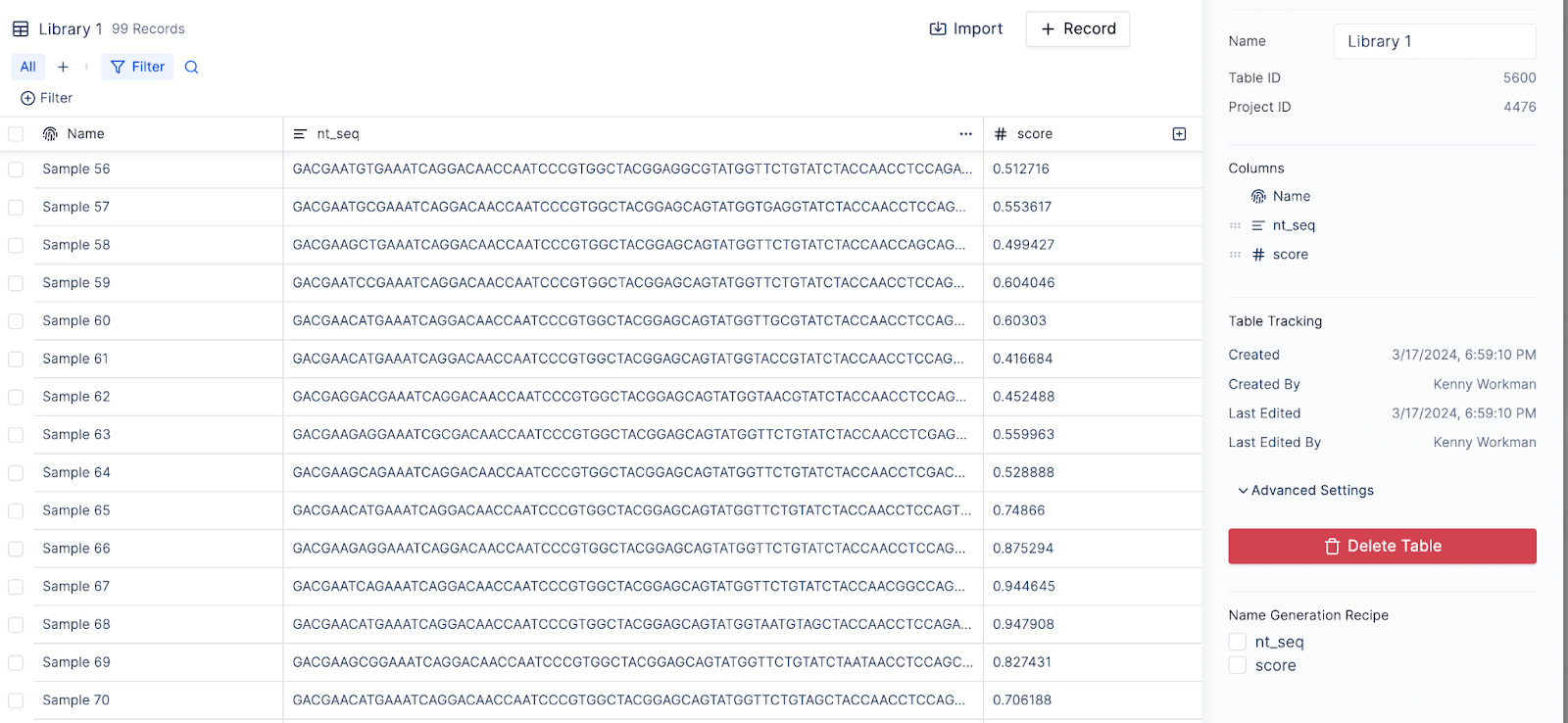
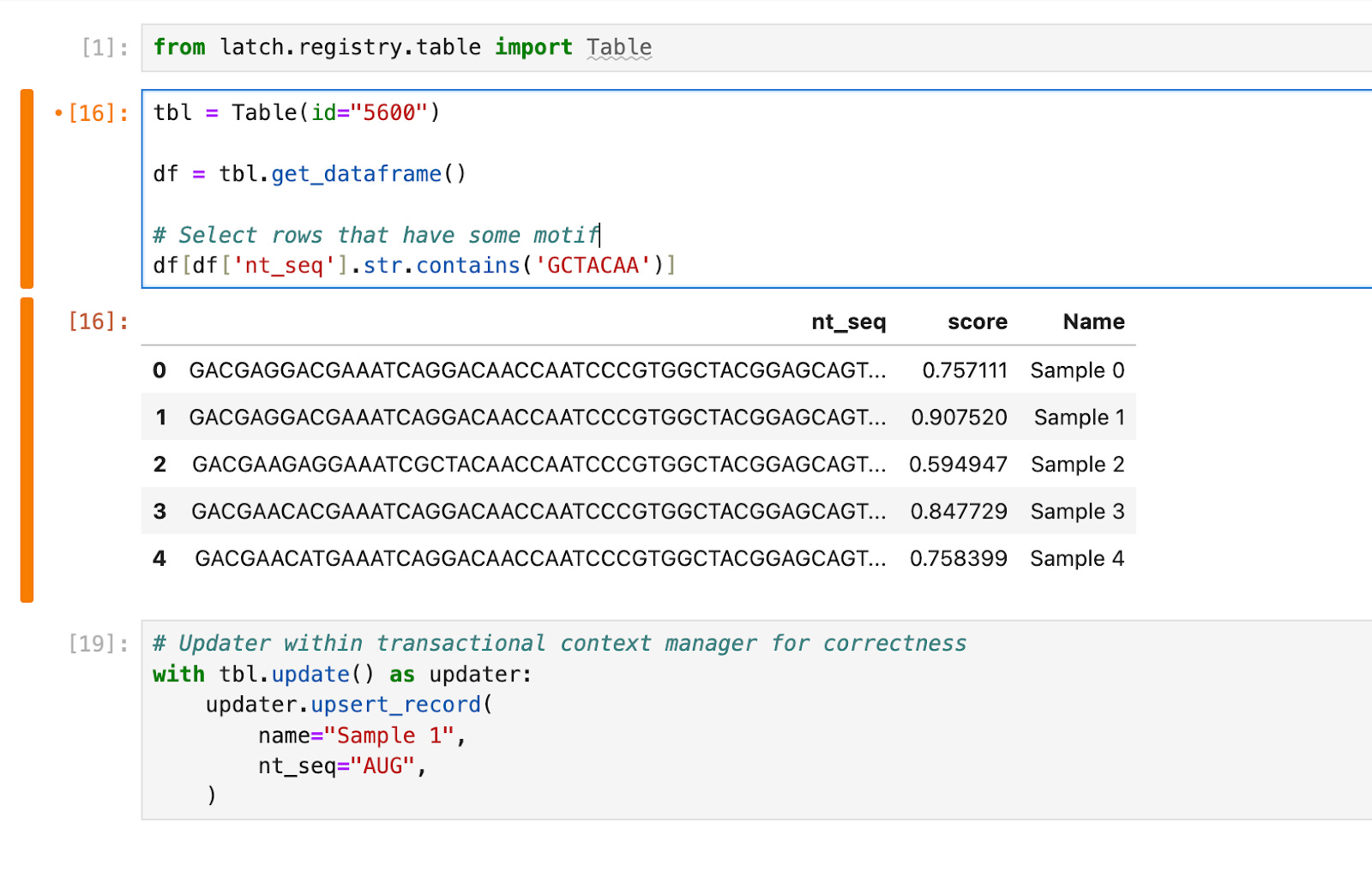
The database needs to be accessible to downstream teams with different scientific backgrounds. In our AAV example, a structural biology team might first want to run computational analysis before passing a subset of the library to a molecular biology team for synthesis and cloning. The structural biologist would search, analyze and write sequences to the shared database using Python in a Jupyter notebook. The API allows the manipulation of tables with pandas Dataframes and ensures that writes back to the database are transactional and correct.
The molecular biologist needs no-code tools to search, filter and download tables to prepare their oligo orders. Building trust amongst scientific teams with new tools is difficult, and the interface needs to be as easy to use and familiar enough to the ubiquitous Google Sheets / Excel to drive adoption, while providing new functionality like table views, CSV import and integrations to popular LIMS.

The ideal database would meet both groups of users at their level of computational fluency, allowing the entire organization to use the same source of truth and spend more time drawing conclusions from the data. These libraries can also be hundreds of thousands or even millions of sequences in size. Both the graphical interface and the programmatic API need to facilitate search and manipulation of database tables with the same speed and UX/DX as the scale of data increases. This is a nontrivial software engineering endeavor.
Validating candidates in-silico with batched AlphaFold2
One method to evaluate the viability of our Evo-generated capsids is to compare the predicted protein structure with the crystallography structure of the wild type sequence and search for obviously malformed features, either visually or programmatically. A structural biology team can then filter out pathological structures from the candidate designs to reduce the resources needed to synthesize and test the full library.
Running AlphaFold2 at this scale (1e4-1e5 runs) requires an orchestration engine to spin up large clusters of computers, dispatch containerized environments, stage file data, store logs and track the execution lifecycle. The batched runs read from and write to our database tables directly, to encourage centralization, organization and correctness of the filtered capsids. It is a waste of resources for downstream molecular biology teams to have to track down sheets of sequences or wrangle inconsistently formatted files; a larger waste if they end up ordering the incorrect designs.

Predicted structures can be inspected directly in the database interface, where linked .pdb files can be located and viewed in a shared filesystem (by clicking on the database cell with the file value). A handful of structures can be inspected this way.

(VP1 fragment variant generated from Evo and folded with AlphaFold)
In the post AlphaFold era, the accessibility of structural models has led to a growing collection of programmatic methods to analyze structure and infer function. To screen a large batch of structures, computational biologists with a bit of Python experience can query the database in a Jupyter notebook, read the necessary .pdb files over a shared networked filesystem and analyze structures at scale. Here we use DSSP to assign secondary structure to sections of the peptide using coordinate information.

Placing orders for libraries of oligos
After computational biologists have finished filtering candidate sequences and performing downstream structural analysis, the designs are passed to the molecular biology team to order synthetic oligos and clone them into a library of plasmids.
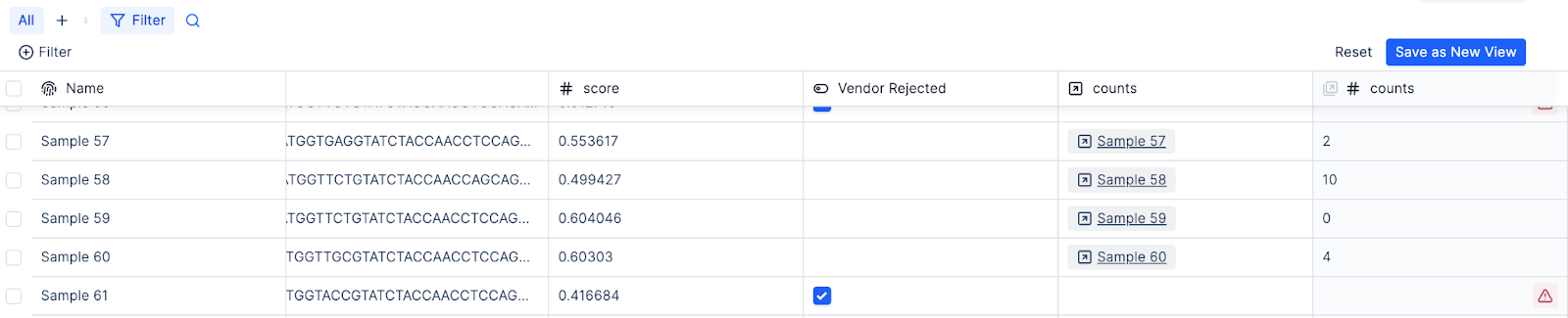
Some of the sequences are rejected by the synthesis vendor due to tricky repetitive regions meddling with “modern” phosphoramidite chemistry used by one of these well branded players (get here faster Ansa, Avery and friends). The molecular biologists return to the database, create a new column and check a box for a handful of rejected sequences.

Analyzing functional validation assays
The shipment of DNA comes in, and after a good deal of cloning, molecular biologists transduce capsid expressing plasmids into plates of cell culture and induce expression. The cells grow and the culture is sequenced. Viable capsid designs will correctly assemble and carry their own genetic cargo, which will be recovered with sequencing and bioinformatics, while unviable designs will be absent. A table of sequencing read counts can be constructed for the library of designs, where high counts indicate viability.
Once again, data must exchange hands between different teams. After prepping their samples and submitting them for sequencing, the molecular biology team creates a sample sheet within the shared database to record experimental metadata (eg. treatment condition, growing temperature), sample identifiers and location in the shared filesystem where sequencing data will be uploaded.
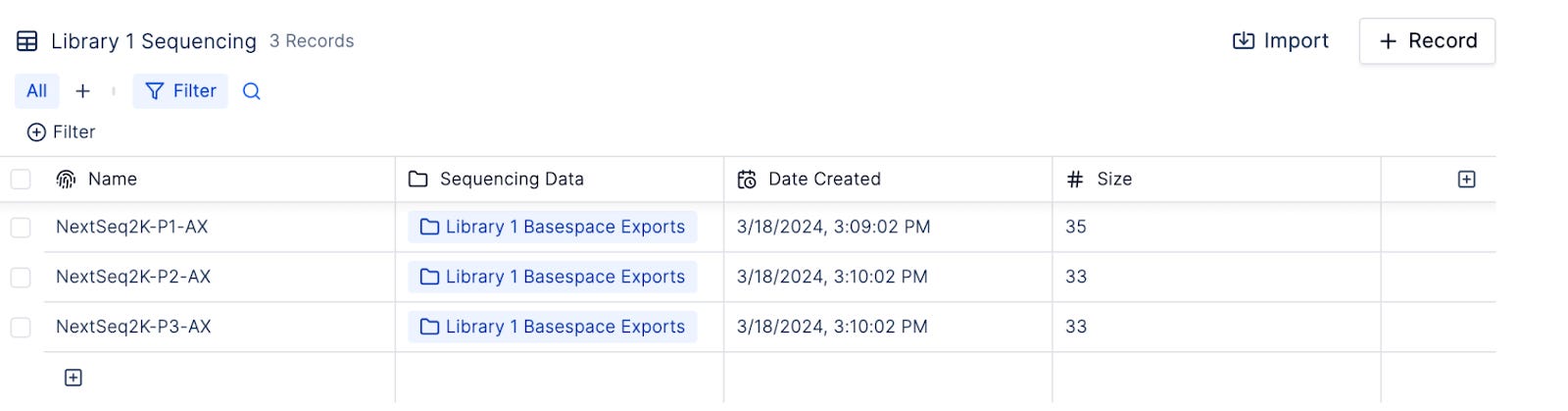
When sequencing completes, data streams from the sequencer automatically to the shared filesystem.

A bioinformatics team has prepared a workflow to process the sequencing data and recover counts for the library of capsids. They program in their language of choice (eg. Python, snakemake or nextflow) and register their code with Latch to generate a graphical interface for scientists to use:
metadata = LatchMetadata(
display_name="vp1_counter",
author=LatchAuthor(
name="Kenneth",
),
parameters={
"fastq_f": LatchParameter(
display_name="Input File"
),
"output_dir": LatchParameter(
display_name="Output Directory"
),
},
)
@workflow(metadata)
def vp1_counter_wf(
fastq_f: LatchFile, output_dir: LatchOutputDir
) -> LatchFile:
return vp1_counter_task(fastq_f=fastq_f, output_dir=output_dir)The bioinformaticians are able to repurpose Fulcrum Genomics’ Rust-based guide-counter, extending the tool to count capsid sequences rather than CRISPR guides, and handling bespoke details of the viability assay, such as custom barcode error correction and UMI deduplication (code for a LatchBio implementation of Fulcrum’s guide-counter available here).. With control over the code, they can iterate in a tight loop with wet lab users.

However, there is no need for a human in the loop as a batch of the sequencing workflows are launched automatically when NGS files are uploaded to the shared filesystem.

The sequencing workflows write directly back to the shared database and add a column of counts values next to the original capsid sequences. These counts represent experimentally validated viability and effectively close our loop.
Closing the loop - drawing meaningful biological conclusions
With experimental data structured and centralized, we are able to proceed by answering simple biological questions.
Relational table joins can be constructed graphically to allow us to associate experimental metadata with processed counts and capsid sequences.
Wet lab teams can also access and explore count data directly by constructing plots from tables. Hovering over plot features reveals metadata from the table, allowing teams to easily identify high performing constructs for next steps. All of the data and assets needed to reproduce the figure are stored in a single place, so modifications can be made easily without tracking down code and raw data from an interesting plot can always be sourced for further analysis at any point in the future.

Teams can answer interesting questions, like how predictive Evo’s log likelihood scores are of capsid viability. We can observe a strong correlation.
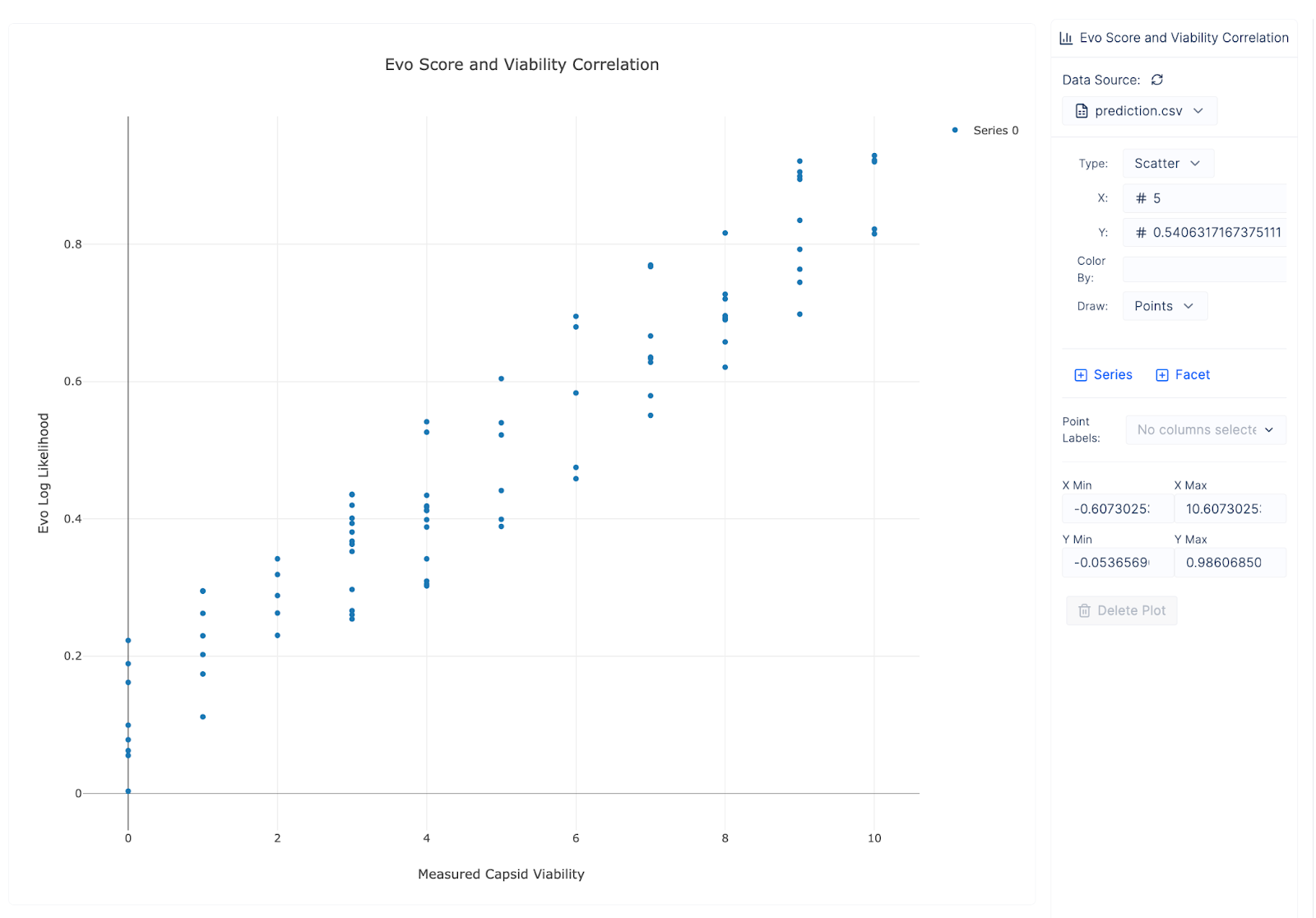
Or how distributions in count data change over multiple iterations of the loop. Our system seems to do its job and each successive round of experimentation increases the average viability of our capsids (different colors represent distinct rounds):
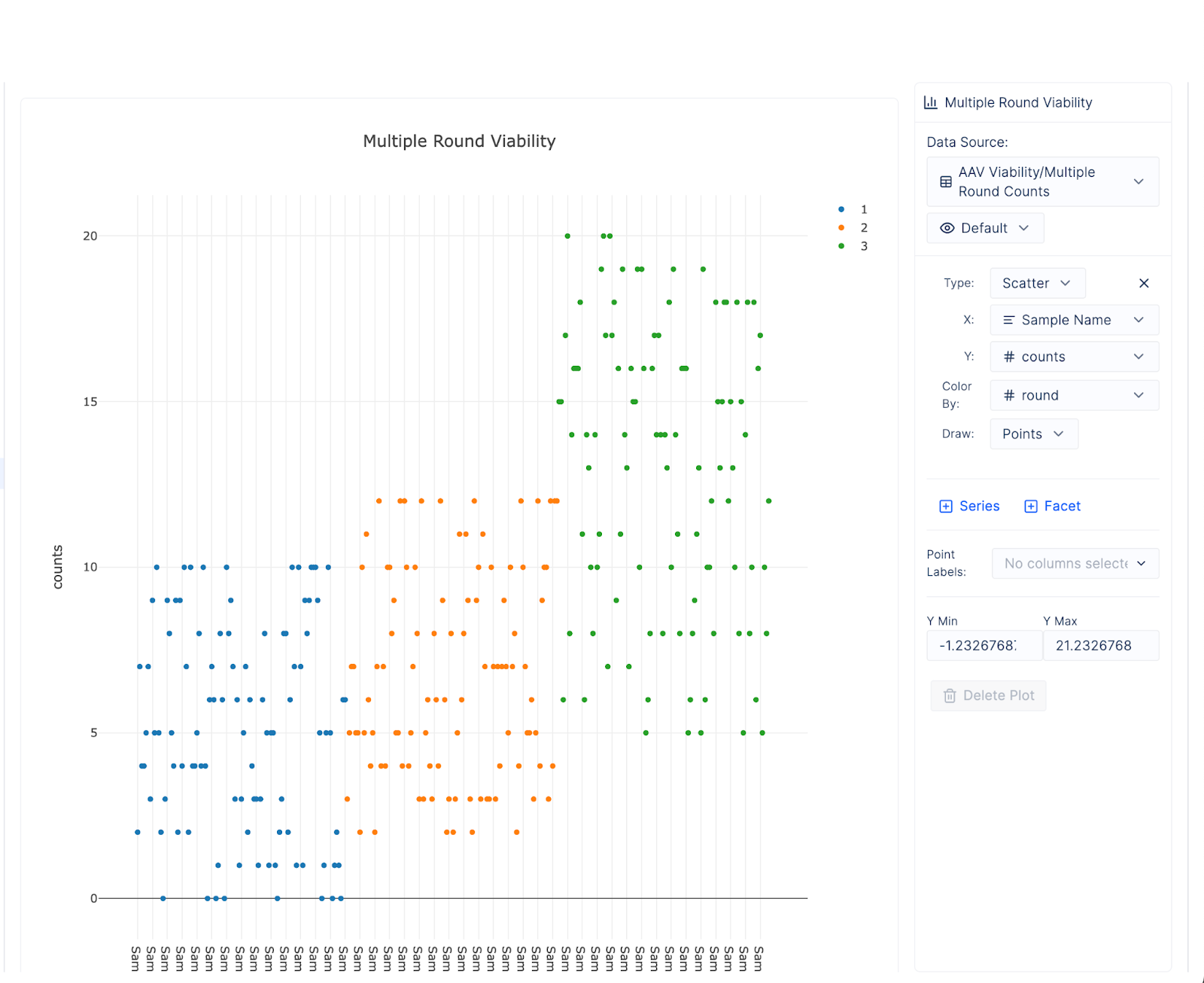
Many of the challenges in preparing these tables for basic analysis - consolidating different types of experiments, structuring input from multiple teams, ingesting data from large computers - have been alleviated by using a single platform for the entire duration of the loop. We are left with simple and accessible tables, where each row is a distillation of immense organizational and technical complexity, but allow everyone at the biotech to explore the raw data and draw their own scientific conclusions.
To Conclude
The modern biotech uses machine learning, distributed computing, metadata management software and high throughput molecular screening to engineer living systems. These different streams of experimental data and new challenges in interdisciplinary coordination are transforming biotech into an industry of information management.

A data platform like LatchBio is a window into the real-time state of biology across experiments and teams that tames this complexity. It provides traceability and accessibility of every spreadsheet from every machine and reproducibility of every analysis. It integrates disparate modalities and scales computational infrastructure to support the ebs and flows of experimental biology. Gold standard machine learning models and bioinformatics tools, from the cutting-edge of computational research, are available for the industry with best practice defaults. Most importantly, all teams have a single source of truth for their data, in a format that suits their unique background.
We hope the industry adopts data platforms. With them we can tackle greater bioengineering challenges and mold life in our image.

Resources
Reach out to install the LatchBio platform at your organization here
Book a 1-on-1 personalized demo with a bioinformatics engineer to learn how your science and infrastructure needs map onto Latch.
Join our upcoming webinar on March 21 with Elsie Biotechnologies how they reduced speed of oligonucleotide therapeutics target discovery by 6 months, formed early partnerships with GSK, and conducted tech transfer with LatchBio’s platform.
Acknowledgements
Thank you to Brian Naughton, Aidan Abdulali, Brandon White, Hannah Le, Patrick Hsu, Kyle Giffin, Alfredo Andere, Tess Van Stekelenburg, Emma Krivoshein, Jordan Ramsay, Blanchard Kenfack for comments and feedback; Nathan Manske for the designs and pictures.

Citations
[1] Bryant, D.H., Bashir, A., Sinai, S. et al. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 39, 691–696 (2021). https://doi.org/10.1038/s41587-020-00793-4
[2] Pierce J. Ogden et al., Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design.Science366,1139-1143(2019).DOI:10.1126/science.aaw2900
[3] Eric Nguyen, Michael Poli, Matthew G. Durrant, Armin W. Thomas, Brian Kang, Jeremy Sullivan, Madelena Y. Ng, Ashley Lewis, Aman Patel, Aaron Lou, Stefano Ermon, Stephen A. Baccus, Tina Hernandez-Boussard, Christopher Ré, Patrick D. Hsu, Brian L. Hie
bioRxiv 2024.02.27.582234; doi: https://doi.org/10.1101/2024.02.27.582234